Dosis respuesta de diferentes inhibidores para estudios metabólicos en astrocitos con L-lactato en concentraciones perinatales.
Jairo Alfonso Tovar Franco MSc, PhD.
Departamento de Nutrición y Bioquímica, Pontificia Universidad Javeriana.
Recibido: 02-05-2005 Aceptado: 20-05-2005
Los sistemas de transporte como las lanzaderas mitocondrial/citosólicas son importantes para las funciones integrativas del cerebro en desarrollo. Para facilitar su estudio y conocer el papel que estos sistemas de transporte puedan tener en células cerebrales se ha empleado trasadores radiactivos en cultivos primarios. En este trabajo se evaluaron las velocidades de oxidación (respiración) y lipogénesis utilizando lactato que es el principal sustrato cerebral durante la prelactancia en astrocitos en cultivo primario. Se determinó las concentraciones mínimas de inhibidores que interactúan con las principales lanzaderas de manera que no fueran tóxicos para las células, pero que su vez permitieran observar efectos metabólicos. Se utilizó L-[U-14C]- lactato (1 MCi), L-lactato (10.5 mM) en ausencia y presencia de diferentes concentraciones de inhibidores enzimáticos como el dicloroacetato (DCA), un inhibidor de la piruvato deshidrogenasa quinasa y el aminooxiacetato (AOA), un inhibidor de la aspartato aminotransferasa e inhibidores del transporte mitocondrial como el butilmalonato (BM), un inhibidor del transporte de dicarboxilatos y específicamente del L-malato y el 1,2,3-benceno tricarboxilato (BT) un inhibidor del transporte de tricarboxilatos. Los resultados sugieren que a concentraciones de 1 mM de DCA y de 5 mM de AOA, BM y BT son suficientes para evaluar efectos metabólicos en células cerebrales durante el periodo perinatal.
Palabras claves: astrocito, inhibidor, lipogénesis, metabolismo, respiración.
The transport systems such as the mitochondrial/cytosolic shuttles are important for the integrating functions of the brain in development. To facilitate their study and to know the rol that these transport systems could have in cerebral cells, radioactive tracers in primary cultured have been used. In this work the oxidation (respiration) and lipogenic rates were evaluated using lactate which is the main cerebral substrate during the presucking in astrocytes in primary cultured, being focused in determinating the minimal inhibitors concentrations that interact with the main shuttles without toxic effects on tested cells; these concentrations must allow us to observe metabolic effects at the same time. Uses of L-[U-14C]-lactato (1 MCi), L-lactato (10.5 mM) in absence and presence of different concentrations of enzymatic inhibitors as dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase and aminooxyacetate (AOA), an inhibitor of aspartate aminotransferase and inhibitors of the mitochondrial transport like butylmalonate (BM), an inhibitor of the dicarboxilic acid transport and specifically of L-malate and the 1,2,3-benzene tricarboxylate (BT) an inhibitor of the tricarboxilic acid transport. The results suggest that concentrations of 1 mM of DCA and of 5 mM of AOA, BM and BT are enough to evaluate the metabolic effects in cerebral cells during the perinatal period.
Key words: astrocyte, inhibitor, lipogenic, metabolism, respiration.
Resultados obtenidos en cortes de cerebro, células cerebrales aisladas, así como en neuronas y astrocitos en cultivo primario han demostrado que el lactato es un extraordinario sustrato cerebral alternativo durante el período perinatal (1-5), siendo utilizado como metabolito energético y precursor lipogénico inmediatamente después del nacimiento (6-8). Las elevadas concentraciones de lactato plasmático (8-10,5 mM), así como su rápido consumo durante las dos primeras horas de vida extrauterina, sugieren que este sustrato satisface las necesidades energéticas del cerebro en este período (9).
Además de servir como precursor de piruvato, el lactato es utilizado por los astrocitos para exportarlo hacia las neuronas (10, 11). En la mitocondria astrocitica, tanto el piruvato como el acetil-CoA, sustrato y producto principales del complejo piruvato deshidrogenasa (PDHC), ocupan una posición clave en el metabolismo al constituir encrucijadas de vías metabólicas. El PDHC se inactiva por fosforilación y se activa por defosforilación. Estas modificaciones son catalizadas, respectivamente, por una piruvato deshidrogenasa quinasa y una piruvato deshidrogenasa fosfatasa. Se sabe que el dicloroacetato (DCA) es un activador del PDHC, a través de la inhibición de la piruvato deshidrogenasa quinasa, la enzima que fosforila la subunidad a del complejo. Por tanto, se quiso estudiar la importancia de la activación del PDHC, haciendo una serie de experimentos en presencia de DCA, usando cultivos de astrocitos.
Por otro lado, en el cerebro en desarrollo existen varios sistemas para el transporte de carbonos de la mitocondria al citosol tales como el transportador de tricarboxilatos responsable del transporte de citrato, cis-aconitato e isocitrato; el transportador de dicarboxilatos responsable del transporte de L-malato, succinato y también de citrato; y la lanzadera del N-acetil-L-aspartato, responsable del transporte de acetil-CoA y aspartato en la forma de NAA. Durante el período prenatal se puede estudiar la relación funcional entre las oxidaciones NAD+ exógenas y endógenas unidas a L-malato y las deshidrogenases NADH dependientes mitocondriales y el efecto sobre el transporte de citrato al citosol como precursor lipogenico, utilizando inhibidores como el aminooxiacetato (AOA), un inhibidor de la aspartato aminotransferasa, el butilmalonato (BM), un inhibidor del transporte de dicarboxilatos y específicamente del L-malato y el 1,2,3-benceno tricarboxilato (BT) un inhibidor del transporte de tricarboxilatos.
Con el empleo de sustancias que intervienen en el metabolismo y en el transporte, especialmente de algunos de los intermediarios de la glucólisis y el ciclo de los ácidos tricarboxílicos (TCA) en astrocitos en cultivo primario se busca tratar de interpretar la compartimentación complicada durante el desarrollo del cerebro y entender cómo se afecta la homeostasis en general y la lipogénesis en particular de este tipo de células. El empleo de inhibidores metabólicos y del transporte han permitido evaluar la actividad de algunas vías durante periodos específicos del desarrollo en células específicas, sin embargo, hay que buscar la concentración mínima de este tipo de sustancias que active o inhiba un proceso sin inducir necrosis o apoptosis. En este sentido, la primera aproximación para estudios metabólicos in vitro, es determinar las concentraciones mínimas de inhibidores claves del transporte mitocondrial e inhibidores enzimáticos que permitan observar alteraciones en las vías oxidativa y lipogénica.
Para conocer el efecto sobre el metabolismo del lactato, astrocitos de 13 días en cultivo, se incubaron a 37°C durante una hora, en tampón fosfato a pH 7.4 (Elliot, 1969), que contiene L-lactato (10.5 mM) y L- [U-14C]-lactato (1 µCi)(100-500 dpm/nmol). Para estudiar el efecto sobre el funcionamiento de las lanzaderas de carbono, es decir, aquellos mecanismos de transporte de carbonos entre la mitocondria y el citosol, se determinaron las velocidades de oxidación (respiración) y lipogénesis en presencia de concentraciones variables de inhibidores enzimáticos e inhibidores del transporte.
Composición del medio Elliott de incubación.
Esta solución tiene una composición aproximada a la del fluido cerebro espinal. Tiene más baja concentración de calcio que el medio Krebs y Henseleit, pero aproximadamente la misma cantidad de calcio ionizado en el plasma. El calcio tiende a precipitar con el fosfato, para asegurarse que no precipita, se adiciona una solución que tenga como máximo una concentración de 11 mM en fosfato de sodio (18). La composición definitiva fue: NaCl (122 mM), KCl (4.8 mM), KH2PO4 (0.4 mM), MgSO4 (1.2 mM), CaCl2 (1.3 mM), preparado en tampón fosfato sódico 10.8 mM a pH=7.6. Extemporáneamente, el pH se ajustó a 7.4, se filtró (0.22 µm poro) y se gaseó con oxígeno durante dos horas (292.4 mOsm/kg H2O).
Preparación de las soluciones para las incubaciones. Se ha elegido, de modo general, concentraciones saturantes de L-lactato (10.5 mM) según trabajos previos (1-3). Se hizo determinación cuantitativa del substrato frío en el medio previamente a la incubación (19) así como para calcular la actividad específica (dpm/nmol). Los inhibidores empleados fueron: ácido dicloroacético (DCA) (CHCl2COOH), ácido aminooxiacético (AOA) (H2NCOCOOH), ácido butilmalónico (BM) [CH3(CH2)3CH(COOH)2] y ácido 1,2,3 - bencenotricarboxílico (BT) hidratado (C9H6O6.H2O). Se tuvo en cuenta las concentraciones de estos inhibidores para ajustar la osmolaridad a valores cercanos a 315 mOsm/kg H2O. A 1.5 ml de medio de incubación Elliott, con los respectivos sustratos fríos por frasco Roux, se adicionaron, de acuerdo al experimento, los trazadores radiactivos con y sin inhibidor. De cada solución se tomaron 100 µL para determinar la radiactividad inicial necesaria para calcular la actividad específica (dpm/nmol).
Incubación con cultivos primarios de astrocitos.
Se emplearon astrocitos de 13 días en cultivo, que habían crecido en frascos tipo Roux. Después de retirar el medio de cultivo se lavaron los frascos 2 veces con PBS y se añadió el medio de incubación (18), con los substratos fríos y radiactivos deseados sin y con el inhibidor. Para ello se incuban a 37°C durante una hora en tampón fosfato a pH 7.4 (Elliott, 1969) que contiene: L-lactato (10.5 mM), L-[U-14C]-lactato (100-500 dpm/nmol), en ausencia o presencia del inhibidor. Se emplearon las siguientes concentraciones: para dicloroacetato (5 µM, 25 µM, 50 µM, 500µMó 1000 µM); aminooxiacetato (5 mM ó 10 mM); butilmalonato (5 mM ó 10 mM);1,2,3- bencenotricarboxilato (5mMó 10 mM).
Posteriormente se gasearon los frascos con O2 durante 30 segundos, se cerraron herméticamente con un tapón de goma y se incubaron a 37°C. En paralelo se llevaron frascos sin células pero con medio de incubación y sustratos fríos y radiactivos. Estos frascos sirvieron de blanco. Para finalizar la incubación los frascos se enfriaron a 4°C (2, 3).
Cuantificación de CO2. Para capturar el 14CO2 producido durante la incubación se empleó el método descrito por Sykes, con algunas modificaciones (20, 21). Para capturar el CO2 se utilizó un matraz Erlenmeyer con un pocillo central que soportaba un eppendorf con 500 µl de hidróxido de hiamina, que se utiliza para atrapar el CO2 en los experimentos de respiración y tiene como característica la de emitir quimioluminiscencia de baja energía (apantallamiento). En el pocillo principal se adicionó 100 µl de KOH (10 M) después de lo cual se selló herméticamente con un tapón de goma.
Sin destapar el frasco de cultivo, se extrajo con una jeringuilla el medio de incubación y se inyectó en el correspondiente matraz erlenmeyer previamente preparado. Se lavó la monocapa de células con PBS que posteriormente fue recuperado en su respectivo matraz. Se inyectaron 2 ml de KOH (0.3 M), colocando el frasco de manera que el KOH no contacte con las células pero capture el CO2 remanente en el frasco de cultivo. Una hora después, el KOH fue retirado e inyectado en su respectivo matraz erlenmeyer. Se realizó otro lavado con PBS y, por último, una vez reunidos todos los volúmenes se adicionó 100 µl de HClO4 (5 M) en el matraz erlenmeyer, con objeto de acidificar el medio y volatilizar el 14CO2 que luego es capturado por la hiamina, este proceso dura 1 hora.
A continuación se recogieron todos los tubos Eppendorf que contienen el hidróxido de hiamina, se colocaron en viales y se les adicionó 5 ml de líquido de centelleo, se agitaron por 30 segundos y se dejaron en reposo 16 horas para medir las desintegraciones por minuto (dpm) utilizando la técnica de espectroscopia de centelleo líquido. La velocidad de respiración se reporta como nmol CO2 /hora /millón de células.
Cuantificación de lípidos. Para hacer la determinación de la incorporación de los sustratos en lípidos totales, se siguió el método recomendado para el aislamiento y purificación de lípidos (22), con ligeras modificaciones (1, 2, 7). Para extraer los lípidos, la monocapa de células se separó del frasco de cultivo con ayuda de un raspador en 1 ml de metanol, y se añadió a un tubo que contiene 2 ml de cloroformo (bidestilado). El tubo se agitó por 30 segundos y se almacenó durante 16 horas en el congelador. Posteriormente se centrifugó (1500 x g, 15 min, 4°C) y se lavó con NaCl (0.3%) saturado con cloroformo. Se realizó una centrifugación en las condiciones anteriores, se retiró la fase acuosa y se recogió la fase clorofórmica. Esta última fase se evapora y el residuo lipídico se disolvió en líquido de centelleo, se agit ó mecánicamente por 30 segundos y después de 24 horas, se midió la radiactividad incorporada en lípidos utilizando la técnica de espectroscopia de centelleo líquido. La velocidad de lipogénesis se reporta como nmol de lípidos/hora /millón de células.
Cálculo de la velocidad de utilización de sustratos. La velocidad de utilización de los substratos se calcula dividiendo la radiactividad incorporada en CO2 o lípidos, por la radiactividad específica del sustrato en el medio de incubación (dpm/nmol), por el tiempo de incubación (1 h) y por el número de células (x 106) (1). A las dpm de oxidación se les resta el valor del blanco. De acuerdo con este cálculo, los resultados se expresan como nmol de sustrato radiactivo transformados en CO2 o en lípidos, por hora y por 106 células.
Determinaciones radiométricas. Las determinaciones de radiactividad se realizaron utilizando un contador centelleo líquido (Beckman). La corrección del apantallamiento de las muestras en disolución acuosa se hizo empleando cantidades crecientes de hidróxido de hiamina (de 0 a 500 µL) como sustancia apantallante. El cálculo del apantallamiento en las muestras se realizó por interpolación en la curva de eficiencias del contaje, ajustada a una ecuación polinómica de tercer grado. La eficiencia del aparato se calculó en un 96-97% para la curva para la radiactividad inicial y los blancos; del 90-92% para la curva de 14CO2 y del 85-86% para la curva de lípidos totales (7, 19, 25).
Tratamiento estadístico. Los ensayos se condujeron según un diseño completamente aleatorizado, con triplicado, como mínimo, de cada determinación. El análisis de la significatividad entre las réplicas de un mismo experimento se ha efectuado utilizando el test «t Student». Los resultados obtenidos fueron evaluados estadísticamente usando el programa para Macintosh, STAT.VIEW. Se hicieron comparaciones múltiples utilizando el análisis de varianza (ANOVA), para determinar los efectos de los diferentes factores considerados. Este análisis incluye el test F de significatividad. Los resultados se expresan como promedios ± la desviación estándar de la media (SEM).
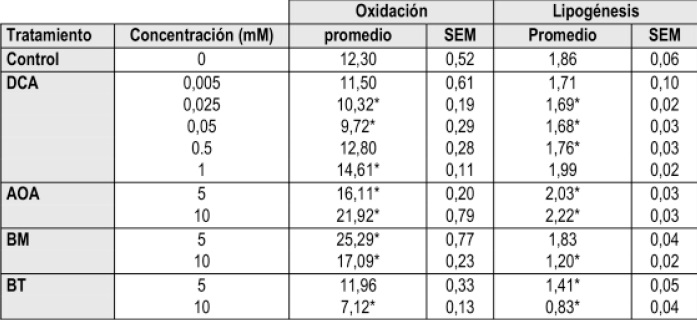
El efecto de diferentes inhibidores sobre la capacidad de utilización de L-lactato en astrocitos se muestra en la tabla 1 y figuras 1 y 2.
Dosis respuesta del dicloroacetato (DCA).
La acción del dicloroacetato (DCA) sobre la actividad del complejo piruvato deshidrogenasa (PDHC) ha sido estudiada en cerebro de ratas in vivo e in vitro. El DCA es un inhibidor de la piruvato deshidrogenasa quinasa y por lo tanto, incrementa la actividad del complejo por disminución de la fosforilación endógena en el cerebro y otros tejidos in vitro (23, 24).
La actividad de la piruvato deshidrogenasa quinasa es mayor en cerebros jóvenes y presenta diferencias en las diferentes regiones cerebrales (25). Así que el DCA, puede inducir cambios en el metabolismo de la glucosa, el lactato y pH en el cerebro (26). También, el DCA inhibe la oxidación del acetato y del 3- hidroxibutirato y parcialmente los efectos inhibitorios reversos de estos substratos sobre la oxidación de glucosa.
Tabla 1. Efecto de concentraciones crecientes de inhibidores enzimáticos y del transporte sobre la utilización del L-lactato por astrocitos en cultivo primario. Astrocitos quiescentes de 13 días se incuban a 37°C durante una hora en tampón fosfato a pH 7,4 que contiene L-lactato (10,5 mM), L-[U-14C]-lactato (1 MCi) en ausencia (control) o presencia de diferentes concentraciones de dicloroacetato (DCA), aminooxiacetato (AOA), butilmalonato (BM) y 1,2,3-benceno tricarboxilato (BT). Las velocidades de oxidación y lipogénesis se expresan como nmol de sustrato transformado en CO2 o lípidos/hora por millón de células y son medias ± SEM. Valores con asterisco (*) presentan diferencias significativas (p<0.01) con respecto al tratamiento control (C).
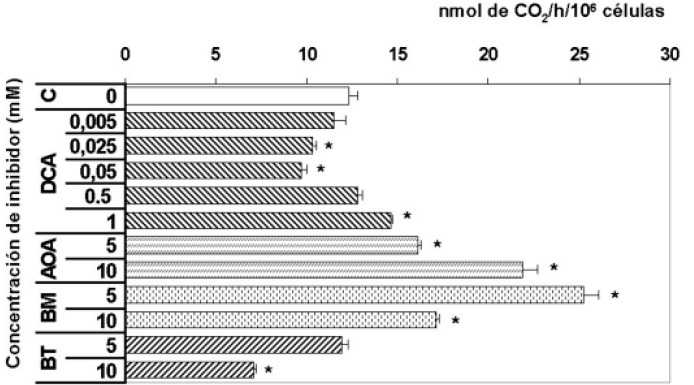
Figura 1. Comparación del efecto de los inhibidores sobre la oxidación. C = control, DCA = dicloroacetato, AOA = aminooxiacetato; BM = butilmalonato; BT = 1,2,3-benceno tricarboxilato.
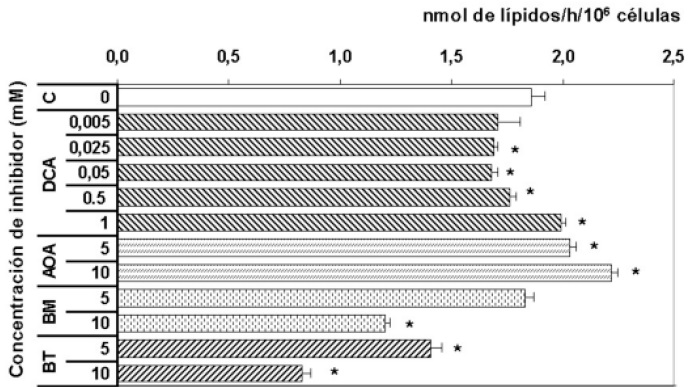
Figura 2. Comparación del efecto de los inhibidores sobre la lipogénesis. C = control, DCA = icloroacetato, AOA = aminooxiacetato; BM = butilmalonato; BT = 1,2,3-benceno tricarboxilato.
El acetil-CoA, acetilcarnitina y glutamato incrementan su concentración celular, mientras aspartato y malato disminuyen por la presencia de este inhibidor (27). La PDH es un enzima clave para el metabolismo del lactato, este metabolismo puede ser facilitado por la activación inducida del DCA sobre el PDHC (26). El DCA puede ser transportado dentro del cerebro por el sistema de transporte de ácidos monocarboxílicos, MCT1 (10, 11, 28). Se ha visto que este compuesto se acumula más rápidamente en cerebros de rata de 20-25 días de vida postnatal que en cerebros de rata adulta, esto debido a la gran capacidad del sistema de transporte durante el período de desarrollo.
En cerebros inmaduros de rata, el DCA incrementa el metabolismo del piruvato y lactato cerebral y como resultado hay un aumento en la actividad glucolítica cerebral (24). Utilizando células de astrocitoma humano de la línea celular UC-11MG se ha demostrado que el DCA no sólo incrementa la actividad del PDHC, sino que reduce la concentraciones de lactato induciendo un eflujo hacia piruvato, además inhibe competitivamente la absorción de lactato (Ki=1.9 mM) (29).
Se ha descrito que el DCA inhibe la piruvato deshidrogenasa quinasa en cortes de cerebro de rata incrementando la actividad del PDHC. Este hecho resulta en la disminución de los niveles de piruvato y lactato, así como estimula la velocidad de utilización de la glucosa (24, 28). Así mismo, el DCA incrementa los niveles de ATP en el cerebro (26) y la acumulación de calcio en mitocondrias del mismo tejido. El DCA puede ser transportado dentro del cerebro por el sistema transportador de ácidos monocarboxílicos que es muy activo en cerebros de rata neonatal (24). No obstante, cruza la barrera hematoencefálica en ratas adultas razonablemente bien (23).
El DCA también inhibe la oxidación del acetato, 3-hidroxibutirato y palmitato lo que sugiere que, el DCA al activar la oxidación de la glucosa a piruvato, aumentaría la competencia por la CoA lo que causaría una disminución de la oxidación de estos otros sustratos al tener enzimas CoA-dependientes. Esto causaría una inhibición de la oxidación de los ácidos grasos que seria reforzada por una disminución de citrato (27).
Ante la posibilidad de que el DCA pudiese tener efectos contrarios a concentraciones elevadas, para buscar la concentración mínima que active la oxidación del piruvato en astrocitos se emplearon cinco concentraciones de dicloroacetato (DCA). La presencia de DCA a concentración de 1 mM aumento significativamente (p<0.01) la velocidad de oxidación y lipogénesis a partir de lactato en astrocitos procedentes de cultivo primario. Es así, como concentraciones más bajas representan en menores velocidades lipogénicas sin mostrar efecto sobre la oxidación de lactato.
El efecto del DCA sobre la oxidación de lactato en astrocitos fue bifásica. En este sentido, concentración de 5 y 25 µMinhiben la oxidación del lactato, mientras que en presencia de concentración de 1 mM de DCA aumenta ligera aunque significativamente (p<0.01) la oxidación del lactato. Este valor es inferior a la Ki reportada (29), de manera que su efecto competitivo sobre la absorción del lactato se ve disminuido. De acuerdo con estos resultados, se recomienda tomar la concentración de 1 mM con objeto de evaluar los efectos del DCA sobre la oxidación y lipogénesis a partir de L-lactato.
Dosis respuesta del aminooxiacetato (AOA).
Inhibidores de las transaminasas han sido utilizados para el estudio de los mecanismos de transporte de aniones a través de la membrana mitocondrial en células intactas. Los primeros estudios han mostrado una fuerte inhibición causada por el AOA en la gluconeogénesis a partir de lactato (30). El AOA es un inhibidor de las enzimas piridoxal-fosfato dependientes en el cerebro tales como, la glutamato descarboxilada y la GABA-B-cetoglutarato transaminasa (31) y se ha usado como un inhibidor del metabolismo del B-aminobutirato (GABA) dado que por inhibe la aminobutirato aminotransferasa (EC 2.6.1.19), previniendo la síntesis de GABA, por lo tanto, se incluye en medios de incubación para inhibir la aminobutirato aminotransferasa previniendo el metabolismo de neurotransmisores y permitiendo el experimento con compuestos marcados.
Asimismo, el AOA es un inhibidor específico de la aspartato aminotransferasa (ASAT) (EC 2.6.1.1), tanto en el compartimento mitocondrial como en el citosólico (32, 33). LaASAT es un componente esencial de la lanzadera malato-aspartato, cuyo papel principal es transportar equivalentes de reducción en forma de NADH desde el compartimento citosólico al mitocondrial (34, 35). En mitocondrias no-sinápticas del cerebro que poseen una activa glutamato-aspartato traslocasa, la oxidación del glutamato es sensible en un 70% al AOA, indicando que este aminoácido es transaminado predominantemente por la ASAT (32, 34, 36). En cortes de cerebro con glucosa como sustrato, la velocidad de consumo de oxígeno disminuye en un 40% por la presencia de AOA mientras que el eflujo a lactato incrementa 3 veces. En estas circunstancias laASAT está inhibida un 97% (31, 37). Por otro lado, la inhibición de la ASAT evitaría la formación del N-acetil-L-aspartato, el segundo aminoácido más abundante en el cerebro (38), que se transporta al compartimento citosólico para proveer los grupos acetilo necesarios para la biosíntesis de los lípidos en la mielinización (39-42).
Un cambio en el potencial redox del citosol, un aumento de la relación lactato/piruvato y una inhibición de la habilidad mitocondrial de utilizar el piruvato glucolítico, son las consecuencias bioenergéticas al adicionarAOA en preparaciones de cerebro, donde es inhibida la lanzadera aspartato-malato previniendo la reoxidación del NADH citosólico por la mitocondria. La relación 3 hidroxibutirato/acetoacetato disminuye significativamente, indicando la oxidación del acople mitocondrial NAD+/NADH (34). Mitocondrias de cerebro no sinápticas poseen una activa glutamato-aspartato translocasa y una oxidación del glutamato que es 70% sensitiva al AOA, indicando que el aminoácido es predominantemente metabolizado por la ASAT. Se ha demostrado que la respiración en cortes de cerebro en presencia de glucosa a disminuido un 40% por el AOA, mientras que el lactato incrementa tres veces (43).
También se ha demostrado que el ß-metilen-Laspartato, un inhibidor selectivo de la aspartato aminotransferasa, tiene un efecto similar, indicando el importante papel de está enzima en el metabolismo de glucosa en el cerebro. Este inhibidor induce una hipoglicemia en terminales nerviosos (34).
En células aisladas de rata neonatal el hidroxicitrato y el AOA disminuyeron la lipogénesis desde lactato en un 40%, sugiriendo que la transferencia de los carbones del lactato de la mitocondria al citosol para la síntesis de lípidos puede estar acoplada a la traslocación de citrato y N-acetil-L-aspartato (4).
Glutamato es el principal neurotransmisor excitatorio en el sistema nervioso central. Uno de los aspectos del metabolismo del glutamato como neurotransmisor es la vía por la cual es sintetizado, las más importantes son las catalizadas por la glutaminasa fosfato-activa, glutamato deshidrogenasa y ASAT. Estudios inmunocitoquímicos han concluido que la ASAT esta asociada con estructuras glutamatergicas. La actividad de ésta enzima en cultivos primarios de gránulos cerebelares es muy alta comparada con el cerebelo.
LaASAT ha sido localizada tanto mitocondrial como citoplasmáticamente e igualmente se ha demostrado que se inhibe la producción de glutamato endógeno cuando las células se exponen a la presencia del AOA y fenilsuccinato. Estas drogas bloquean, respectivamente, laASATy el transporte de ácidos ceto-dicarboxílicos (32).
La inhibición de ASAT disminuiría la acción de la lanzadera malato-aspartato por disminución de las concentraciones de aspartato lo que obligaría a la célula a utilizar otra fuente de este sustrato (30). La adición de AOA causa cambios letales degenerativos en las neuronas, probablemente causados por una toxicidad debida al exceso de glutamato. Igualmente la adición de AOA causa un aumento significativo del contenido de lactato y una disminución de los niveles de ATP (44).
Como puede observarse en la Figura 1 el AOA incremento las velocidades de oxidación y lipogénesis a partir de L-lactato en los astrocitos. La adición de AOA a concentraciones de 5 y 10 mM, aumenta la oxidación a partir de L-lactato en un 26% y 71% respectivamente (p<0.01). Igualmente, la incorporación en lípidos a partir de L-lactato aumenta en un 11 y 21% (p<0.01). Estos resultados indican que al favorecerse el incremento de las concentraciones mitocondriales de oxalacetato, se favoreció un incremento en la respiración de los astrocitos, reflejándose en el incremento de lípidos por estas células. De acuerdo con estos resultados, se recomienda tomar la concentración de 5 mM con objeto de evaluar los efectos del BM sobre la oxidación y lipogénesis a partir de L-lactato.
Dosis respuesta del butilmalonato (BM).
El transportador de citrato es también inhibido por aniones no penetrantes como el butilmalonato (BM) (54% de inhibición), fenilsuccinato, bencilmalonato y pentilmalonato, que son considerados como inhibidores específicos del transporte de dicarboxilatos (30, 45, 46). Con piruvato como sustrato, el eflujo de citrato desde la mitocondria puede ocurrir, en este caso el transporte de malato sirve como fuente de carbonos y de equivalentes de reducción. Con el lactato como sustrato, el transporte de aspartato formado de oxalacetato, al citosol, predomina sobre el transporte de malato, de esta manera los equivalentes reductivos necesarios para la gluconeogénesis son proveídos por la reacción de la lactato deshidrogenasa. Sin embargo, también con el lactato como sustrato, la gluconeogénesis es inhibida por el butilmalonato (BM), bajo estas condiciones el a-cetoglutarato, requerido para la transaminación extramitocondrial con aspartato, fluye continuamente desde la mitocondria al citosol por intercambio con el malato. Este malato, luego, puede ser reciclado desde el citosol presumiblemente por intercambio con fosfato vía el traslocador de dicarboxilatos. Este reciclaje de trazas de malato a través de la membrana mitocondrial a sido demostrado en muchos trabajos. La conclusión es que el BM inhibe la gluconeogénesis por bloqueo del transporte de malato (47, 48). Estudios con mitocondrias aisladas, han mostrado que el intercambio malato-malato a través de la membrana mitocondrial es mediado por tres sistemas de transporte, el traslocador de dicarboxilatos, el traslocador de a-cetoglutarato y el traslocador de tricarboxilatos. De estos traslocadores solamente el primero es sensitivo al butilmalonato (30).
En trabajos previos con células aisladas de cerebro de neonato de rata se hicieron experimentos con concentraciones de BM 5 y 6 mM (3, 4) y hay reportadas en la literatura en estudios con mitocondrias de hígado de rata (5 y 10 mM) (45, 49).
La velocidad de absorción del citrato es inhibida por aniones no penetrantes, tales como el fenilsuccinato, bencilmalonato, pentilmalonato y butilmalonato (BM) a través de la inhibición específica del transportador de dicarboxilatos en mitocondrias aisladas de hígado de rata (45). Un efecto similar se ha encontrado en mitocondrias aisladas de cerebro de rata con el butilmalonato (50). Todos estos análogos dicarboxílicos inhiben el transportador de citrato. Esta inhibición es competitiva. Se ha sugerido que el transportador de tricarboxilatos tiene dos sitios de unión, uno específico para Pi y otro específico para dicarboxilatos. El butilmalonato inhibe un 54% la velocidad de absorción del citrato en mitocondrias de hígado de rata (45). Asimismo, se ha demostrado que el BM es un inhibidor específico del transporte de malato (50).
La adición de 5 mM de BM, aumento significativamente la velocidad de oxidación a partir de L-lactato en un 96%; a concentraciones de BM de 10 mM el aumento fue menor pero aún significativo 33% (p<0.01). No se observo ningún efecto significativo en cuanto a la velocidad de incorporación en lípidos a concentraciones de BM 5 mM; con BM 10 mM se observa una disminución del 37% (p<0.0001).
Estos resultados indican que la inhibición completa de la salida de malato y parcial de citrato de la mitocondria favorecio un incremento en la respiración, sin embargo, el mantenimiento de la lipogénesis a 5 mM y la disminución de tan solo el 35% a concentración de 10 mM del inhibidor pueden estar indicando la existencia de otra ruta alternativa para la síntesis de lípidos desde lactato, como ya ha sido propuesto (5) De acuerdo con estos resultados, se opta por tomar la concentración de 5 mM con objeto de evaluar los efectos del BM sobre la oxidación y lipogénesis a partir de L-lactato.
Dosis respuesta del 1, 2, 3 b enceno tricarboxilato (BT).
El transporte de citrato a través de la membrana interna mitocondrial tambien se inhibe de manera competitiva por algunos tricarboxilatos, tales como cisaconitato, 1,2,3-propano tricarboxilato, propilcitrato y 1,2,3-benceno tricarboxilato (BT) (45).El BT es un inhibidor relativamente específico del transporte del citrato mitocondrial (51). También la oxidación de citrato, cis-aconitato e isocitrato son inhibidas por este compuesto (49). Sin embargo, el BT también inhibe la acetil-CoA carboxilasa inhibiendo la síntesis de ácidos grasos en el compartimento citosólico (30). Las medidas del intercambio de citrato/[14C]citrato; citrato/ L-[14C]malato y malato/[14C]citrato a intervalos de tiempo definidos han confirmado que el BT es un inhibidor de este sistema de transporte, así como análogos del citrato como el 2-etilcitrato y el 2- propilcitrato (45, 49).
El BT es un inhibidor específico del transporte de tricarboxilatos en mitocondrias de hígado de rata (49) así como las procedentes de cerebro de rata (50). Igualmente, se ha demostrado la efectividad del BT sobre el transportador de tricarboxilatos aislado de mitocondrias de hígado de rata e incorporado a liposomas (52). Se ha encontrado, asimismo, que el transportador de tricarboxilatos tiene un sitio simple de unión para los tricarboxilatos, fosfoenol piruvato y dicarboxilatos. Se ha establecido el siguiente orden de afinidad por el transportador de tricarboxilatos: BT (Ki = 0.07 mM) > cis -aconitato (Ki = 0.09 mM) > citrato (Ki = 0.13 mM) > isocitrato (Ki = 0.18 mM) > propilcitrato (Ki = 2.8 mM). En todos los casos el tipo de inhibición es competitivo (45). No obstante, la afinidad del inhibidor de tricarboxilatos por los transportadores de dicarboxilatos es baja.
En células en suspensión de cerebros de rata neonatal el BT y el BM no afectan la velocidad de lipogénesis desde lactato, ni tampoco la oxidación del lactato. El BT no afecta la velocidad de lipogénesis desde 3-hidroxibutirato pero si disminuye ligeramente la integración de los carbonos del 3-hidroxibutirato en lípidos en el citosol, esto puede deberse a que durante el período perinatal el papel del transportador de citrato no es relevante en controlar lipogénesis (4).
Como puede observarse en la tabla 1, la adición de BT 5 mM, no aumento las velocidades de oxidación a partir de L-lactato pero si se aumenta la concentración de BT a 10 mM, se observa una disminución significativa (p<0.01) en la velocidad de oxidación, indicando que el incremento de citrato mitocondrial inhibe el ciclo de los ácidos tricarboxílicos, como es bien conocido. Por otro lado, la velocidad de lipogénesis que disminuyen en 24% con 5 mM y 54% con 10 mM, lo que puede estar indicando un efecto inhibitorio sobre la acetil-CoA carboxilasa (EC 6.4.1.2) en este último caso. Adicionalmente, debido a que el BT posiblemente puede entrar a la mitocondria e inhibir la piruvato carboxilasa (EC 6.4.1.1) (51), una enzima exclusivamente astrocítica (53, 54), lo que explicaría la disminución tan significativa en respiración y lipogénesis cuando se incremento la concentración del inhibidor.
Aun así, la lipogénesis observada solo se podría explicar por la existencia de otra ruta alternativa como ya ha sido propuesto. De acuerdo con estos resultados, se opta por la concentración de 5 mM con objeto de evaluar en cultivos de astrocitos la oxidación y lipogénesis a partir de L-lactato.
La correcta selección de concentraciones de inhibidores para estudios metabólicos, bioquímicos y genéticos no accesibles in vivo permitirá tener una mejor interpretación de los resultados bioquímicos. Se destaca la importancia de contar con cultivos homogéneos de una sola clase de células para investigar mecanismos neuroquímicos, pero al mismo tiempo se recomienda cautela al intentar comparar datos de estos cultivos con los derivados del estudio del cerebro in vivo. Los resultados del presente trabajo se pueden aplicar para estudios metabólicos variando el substrato, el trazador radiactivo o el monocultivo, siendo extensible al estudio de las neuronas en condiciones fisiológicas perinatales.
Al Programa de Cooperación Científica con Iberoamérica en la modalidad de Formación de Investigadores del Ministerio de Educación y Ciencia de España y al Dr. José María Medina, catedrático de la Universidad de Salamanca.
1. Tovar J. Compartimentación celular del metabolismo del lactato en neuronas y astrocitos en cultivo primario. [Tesis doctoral.]. Universidad de Salamanca. 1995. 210 p.
2. Tabernero A. Regulación del metabolismo del lactato en neuronas y astrocitos en cultivo primario. [Tésis doctoral]. Facultad de Farmacia. Departamento de Bioquímica y Biología Molecular. Universidad de Salamanca. 1993. 186 p.
3. Vicario C. Regulación del metabolismo del lactato en células aisladas de cerebro de rata. [Tésis doctoral]. Universidad de Salamanca. Facultad de Farmacia. Departamento de Bioquímica y Biología Molecular. 1991. 263 p.
4. Vicario C, Medina J. Metabolism of lactate in the rat brain during the early neonatal period. J. Neurochem. 1992;59(1):32-40.
5. Medina JM, Tabernero A, Tovar J, Martin-Barrientos J. Metabolic fuel utilization and pyruvate oxidation during the postnatal period. J.Inher.Metab.Dis. 1996;19:432-442.
6. Arizmendi C, Medina J. Lactate as an oxidizable substrate for rat brain in vitro during the perinatal period. Biochem.J. 1983;214:633-635.
7. Bolaños JP. Efecto del valproato sobre el metabolismo intermediario en cerebro de neonato de rata. [Tésis doctoral]. Facultad de Farmacia. Departamento de Bioquímica y Biología Molecular. Universidad de Salamanca. 1992a. 202 p.
8. Fernández E, Medina, JM. Lactate utilization by the neonatal rat brain in vitro. Biochem.J. 1986;234:489-492.
9. Medina JM, Bolaños JP, Vicario C, Arizmendi, C. Fuel supply to the brain during the early postnatal period. In: Cuezva J, ed. Endocrine and biochemical development of the fetus and neonate. New York: Plenum Press; 1996:175-194.
10. Gladden L. Lactate metabolism: a new paradigm for the third millennium. J. Physiol. 2004;558(1):5-30.
11. Tovar J, Saavedra L, Bryon A. Metabolismo cerebral. En: Niño M y Ferrer, L., ed. Neuroanestesia. Enfoque perioperatorio en el paciente neurologico. Bogotá D.C. Distribuna Editorial Médica; 2005:33-88.
12. Cohen J, Wilkin G. Neural cell culture. A practical approach. In: Rickwood D, Hames B. Ed. London, England. IRL press at Oxford University Press. 1995.
13. Saneto R, DeVellis J. Neuronal and glial cells: Cell culture of the central nervous system. In: A.J. Turner & H.S. Bachelard, ed. Neurochemistry a Practical Approach. IRL Press. 1987:27- 64.
14. Rose S, Sinha A. Some properties of isolated neuronal cell fractions. J.Neurochem. 1969;16:1319-1328.
15. Kimelberg HK. Primary astrocyte cultures - A key to astrocyte function. Cell.Molec.Neurobiol. 1983;3(1):1-16.
16. Bigmani A, Eng LF, Dahl D, Uyeda, CT. Localization of the glial fibrillary acidic protein in astrocites by inmunofluorescence. Brain Res. 1972;43:429-435.
17. Kimelberg HK, Norenberg MD. Astrocitos. Investigación y Ciencia. 1989;Junio,:18-27.
18. Elliott KAC. The use of brain slices. In: Laftha A, ed. Handbook of neurochemistry. Vol. 2. New York, NY. Plenum Press. 1969:103-115.
19. Gutmann I, Wilhelm A. L-(+)-Lactate. determination with lactate dehydrogenase and NAD. In: Bergmeyer H, ed. Methods of Enzimatic Analysis. Vol. 3. Deerfiel Beach, FL. Academic Press.; 1974:1464-1468.
20. Edmond E, Robbins R, Bergstrom J, Cole R, De Vellis J. Capacity for substrates utilization in oxidative metabolism by neurons, astrocytes and oligodendrocytes from developing brain in primary culture. J.Neurosci.Res. 1987;18(4):551- 561.
21. Sykes J, López-Cardoso M , Van Den Bergh S. Substrate utilization for energy production and lipid synthesis in oligodendrocyte-enriched cultures prepared from rat brain. Neurochem.Int. 1986;8:67-75.
22. Folch J, Lees M, Slone GH. A simple method for the isolation and purification of total lipides from animal tissues. J.Biol.Chem. 1957;226:497-509.
23. Kuroda Y, Toshima K, Watanabe T, Kobashi H, Ito M, Takeda E, Miyao M. Effects of dichloroacetate on pyruvate metabolism in rat brain in vivo. Pediatric Res. 1984;18:936- 938.
24. Miller AL, Hatch JP, Prihoda TJ. Dichloroacetate increase glucose use and decreases lactate in developing rat brain. Metabolic Brain Disease. 1990;5(4):195-204.
25. Rex-Sheu K-F, Lai JC, Blass JP. Properties and regional distribution of pyruvate dehydrogenase kinase in rat brain. J.Neurochem. 1984;42:230-236.
26. Katayama Y, Welsh FA. Effects of dichloroacetate on regional energy metabolites and pyruvate dehydrogenase activity during ischemia and reperfusion in gerbil brain. J.Neurochem. 1989;52(6):1817-1822.
27. McAllister A, Allison SP, Randle PJ. Effects of dichloroacetate on the metabolism of glucose, pyruvate, acetate, 3- hidroxybutyrate and palmitate in rat diaphragm and heart muscle in vitro and on extraction of glucose, lactate, pyruvate and free fatty acids by dog heart in vivo. Biochem.J. 1973;134:1067-1081.
28. Abemayor E, Kovachich, G, Haugaard, N. Effects of dichloroacetate on brain pyruvate dehydrogenase. J.Neurochem. 1984;42(1):38-42.
29. Tomsig JL, Gruenstein E, Dimlich, RV. Inhibition of lactateinduced swelling by dichoroacetate in human astrocytomacells. Brain Res. 1991;568:92-100.
30. Meijer AJ, Van Dam, K. The metabolic significance of anion transport in mitochondria. Biochim.Biophys.Acta. 1974;346:213-244.
31. Flint M, Swartz KJ, Hyman BT, Storey E, Finn SF, Koroshetz W. Aminooxyacetic acid results in excitotoxin lesions by a novel indirect mechanism. J.Neurochem. 1991;57(3):1068- 1073.
32. Palaiologos G, Hertz L, Schousboe A. Role of aspartate aminotransferase and mitochondrial dicarboxylate transport for release of endogenously and exogenously supplied neurotransmitter in glutamatergic neurons. Neurochem.Res. 1989;14(4):359-366.
33. Yu A, Schousboe, A, Hertz, L. Metabolic fate of 14C-labeled glutamate in astrocytes in primary culture. J.Neurochem. 1982;39:954-960.
34. Kauppinen RA, Sihra T, Nicholls D. Aminooxyacetic acid inhibits the malate-aspartate shuttle in isolated nerve terminals and prevents the mitochondria from utilizing glycolytic substrates. Biochim.Biophys. Acta. 1987;930:173- 178.
35. McKenna M, Tyson J, Couto R, Stevenson J, Caprio F. The metabolism of malate by cultured rat brain astrocytes. Neurochem.Res. 1990;15(12):1211-1220.
36. Farinelli S, Nicklas W. Glutamate metabolism in rat cortical astrocyte cultures. J.Neurochem. 1992;58(5):1905-1915.
37. Passarella S, Barile M, Atlante A, Quagliariello E. Oxaloacetate uptake into rat brain mitochondria and reconstruction of the malate/oxaloacetate shuttle. Biochem.Biophys.Res.Comm. 1984;119:1039 1046.
38. Neale J, Bzdega T, Wroblewska B. N-acetylaspartylglutamate. The most abundant peptide neurotransmitter in the mammalian central nervous system. J.Neurochem. 2000;75(2):443-452.
39. Chakraborty G, Mekala P, Yahya D, Wu G, Ledeen R. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipids synthesis: evidence for myelin-associated aspartoacylase. J.Neurochem. 2001;78:736 745.
40. Patel T, Clark, J. Synthesis of N-acetyl-L-aspartate by rat brain mitochondria and its involvement in mitochondrial/ cytosolic carbon transport. Biochem.J. 1979;184:539-546.
41. D.Adamo A, Smith J, Phillips I. The piruvate dehydrogenase complex: cloning of the rat somatic E1a subunit and its coordinate expression with the mRNAs for E1b, E2, and E3 catalytic subunits in developing rat brain. J.Neurochem. 1973;20:1275-1278.
42. D.Adamo A, Yatsu, F. Acetate metabolism in the nervous system. N-acetyl-L-aspartic acid and the biosynthesis of brain lipids. J.Neurochem. 1966;13:961-965.
43. Fitzpatrick S, Cooper A, Duffy T. Use of ß-methylene-D,Laspartate to assess the role of aspartate aminotransferase in cerebral oxidative metabolism. J.Neurochem. 1983;41:1370- 1383.
44. Beal MF, Swartz KJ, Hyman BT, Storey E, Finn SF, Koroshetz W. Aminooxyacetic acid results in excitotoxin lesions by a novel indirect mechanism. J.Neurochem. 1991;57:1068- 1073.
45. Palmieri F, Stipani I, Quagliariello E. Kinetic study of the tricarboxylate carrier in rat liver mitochondria. Eur.J.Biochem. 1972;26:587-594.
46. Watson JA, Lowenstein, JM. Citrate and the Conversion of Carbohydrate into Fat. Fatty acid synthesis by a combination of cytoplasm and mitochondria. J.Biol.Chem. 1970;245(22):5993-6002.
47. Calvin J, Tubbs P. Mitochondrial transport processes and oxidation of NADH by Hypotonically treated boar spermatozoa. Europ J Biochem. 1978;89:315-320.
48. Coleman J, Palmer JM. The oxidation of malate by isolated plant mitochondria. Eur. J. Biochem. 1972;26:499-509.
49. Robinson BH, Willians GR, Halperin ML, Leznoff CC. The sensitivity of the exchange reactions of tricarboxylate, 2- oxoglutarate and dicarboxylate transporting system of rat liver mitochondria to inhibition by 2-pentylmalonate, piodobenzylmalonate, and benzene 1, 2, 3-tricarboxylate. Eur.J.Biochem. 1971;20(1):65-71.
50. Patel MS. Citrate transport oand oxidation by isolated rat brain mitochondria. Brain Res. 1975;98:607-611.
51. Stucki J. Influence of 1,2,3-benzene-tricarboxylate on pyruvate metabolism in rat-liver mitochondria. Eur J Biochem. 1977;78(1):183-187.
52. Stipani I, Palmieri, F. Purification of the active mitochondrial tricarboxylate carrier by hydroxylapatite chromatography. FEBS. 1983;161:269-274.
53. Patel MS. CO2-Fixing enzymes. In: Boulton AA, Baker GB, Butterworth RF, eds. Neuromethods Carbohydrates and Energy Metabolism. Vol. 11. Clifton, N.J.: Humana Press.; 1989:309-341.
54. Kaufman E, Driscoll B. Evidence for cooperativity between neurons and astroglia in the regulation of CO2 fixation in vitro. Dev.Neurosci. 1993;15:299-305.