Diagnostico de homocistinuria y deficiencia de Adenilosuccinato Liasa a través de técnicas químicas, bioquímicas y moleculares.
Marta Bermúdez PhD.1, Yina Carrillo Bsc.2
1Instituto de Genética Humana Pontificia Universidad Javeriana, Bogotá, Colombia
2Universidad Colegio Mayor de Cundinamarca, Bogotá, Colombia.
Correspondencia: martha.bermudez@javeriana.edu.co .
Recibido: 13-04-2005 / Aceptado:26-05-2005
Entre los errores innatos del metabolismo (EIM) que son defectos bioquímicos de origen genético se encuentra: la homocistinuria y la deficiencia de adenilosuccinato liasa (EC 4.3.2.2) (ADSL), la primera es frecuentemente producida por deficiencia de la cistationina ß sintasa (EC 4.2.1.22) (CßS) enzima que interviene en el catabolismo del aminoácido esencial metionina, la segunda es un defecto en el metabolismo de las purinas. Las manifestaciones clínicas de estas deficiencias son: para la CßS se comprometen sistemas del organismo como el ocular, músculo esquelético, vascular y sistema nervioso central; en el caso de ADSL, se presenta retardo mental, convulsiones, rasgos autistas, movimientos involuntarios, espasmo e hipoplasia cerebral.
En este trabajo se escribe la secuencia utilizada en el diagnóstico de la homocistinuria y de la ADSL, a partir del uso de métodos químicos, bioquímicos y moleculares. Se estudiaron pacientes con sospecha clínica de estar afectados por un EIM; se emplearon las pruebas químicas como el nitroprusiato de sodio y de plata; separación de aminoácidos en plasma y orina por cromatografía de capa fina, técnicas bioquímicas para cuantificar la enzima CßS y técnicas moleculares para identificar la mutación que produce la homocistinuria. En el caso de la ADSL el diagnóstico se realizó por medio del test de Bratton Marshall con el cual se identifica la presencia de metabolitos de las purinas; luego por cromatografía de alta resolución (HPLC) para separar, identifica y cuantifica las succinilpurinas. Para el caso de la homocistinuria los resultados de nitroprusiato de sodio y de plata fueron positivos, la cuantificación enzimática mostró deficiencia de CßS y se llegó a identificar la presencia de una mutación. En el caso de la deficiencia de ADSL el test de Bratton Marshall fue positivo y la cuantificaron de metabolitos de las purinas se encontró aumentada.
Palabras Claves: Cistationina ß sintasa, deficiencia de adenilosuccinato liasa, Homocistinuria, metabolismo purinas, retardo mental, test de Bratton Marshall.
Among the innate errors of the metabolism (EIM) that are biochemical defects of genetic origin is: the homocistinuria and the deficiency of adenilosuccinato liasa (EC 4.3.2.2) (ADSL), the first one is frequently taken place by deficiency of the cistationina ß sintasa (EC 4.2.1.22) (CßS) enzyme that intervenes in the catabolismo of the amino acid essential metionina, the second are a defect in the metabolism of the purinas. The clinical manifestations of these deficiencies are: for the CßS they commit systems of the organism like the ocular, skeletal, vascular muscle and central nervous system; in the case of ADSL, mental retard is presented, convulse, autistic features, involuntary movements, spasm and cerebral hipoplasia. In this work the sequence is written used in the diagnosis of the homocistinuria and of the ADSL, starting from the use of chemical, biochemical and molecular methods.
They were studied patient with suspicion clinic of being affected by a EIM; the chemical tests were used as the nitroprusiato of sodium and of silver; separation of amino acids in plasm and he/she urinates for biochemical fine, technical layer cromatografía to quantify the enzyme CßS and technical molecular to identify the mutation that produces the homocistinuria. In the case of the ADSL the diagnosis was carried out by means of Bratton Marshall test with which the presence of metabolitos of the purinas is identified; then for cromatografía of high resolution (HPLC) to separate, to identify and to quantify succinilpurinas. For the case of the homocistinuria the results of nitroprusiato of sodium and of silver they were positive, the enzymatic quantification showed deficiency of C?S and you ended up identifying the presence of a mutation. In the case of the deficiency of ADSL Bratton Marshall test was positive and they quantified it of metabolitos of the purinas it was increased.
Key words: Cistationina ß sintasa, deficiency of adenilosuccinato liasa, Homocistinuria, metabolism purinas, mental retard, test of Bratton Marshall.
Los EIM son desordenes bioquímicos, de origen genético, ocasionados por un defecto en la estructura de las moléculas proteicas involucradas en alguna vía metabólica, cambio que afecta la función catalítica y como resultados produce acumulación del sustrato y formación insuficiente del producto (1).
Cada día crece el interés del personal médico por obtener información que permita realizar un diagnóstico y tratamiento oportuno de estas patologías. Los EIM individualmente son poco frecuentes pero en conjunto existe una gran diversidad y una creciente y continua descripción de nuevos casos. La mayoría de estas patologías se heredan de forma autosómica recesiva.
Entre estas patologías están la enfermedad de la orina con olor a jarabe de arce (MSUD), acidemias orgánicas, galactosemia, acidosis láctica, ciclo de la urea, trastornos de la ß-oxidacion. Los EIM se pueden clasificar en 5 grupos así: Grupo I: tipo intoxicación: se encuentran tres categorías: a, b y c, según estén o no presentes diversos grados de deshidratación, cetoacidosis e hiperamonemia.
Los tres comparten una clínica neurológica progresiva y severa. Un ejemplo de este tipo es la enfermedad de la orina con olor a jarabe de arce. El Grupo II: tipo déficit energético: se subdivide en a y b, según predomine la disfunción hepática o el binomio hipotonía-polipnea; en la clínica lo más relevante son los síntomas neuromusculares. Un ejemplo son los trastornos en la B-oxidación y la acidosis láctica. El Grupo III: tipo hepatomegaliahipoglucemia: se clasifica en a y b, según el síndrome sea único o asociado a ictericia y afectación hepática preferentemente, el grupo III b se caracteriza por la ausencia de alteraciones bioquímicas. El Grupo IV: predomina las convulsiones en este grupo se puede clasificar la deficiencia de adenilosuccinato liasa. El Grupo V: predomina la tendencia tesaurismótica dentro de este grupo se encuentra la homocistinuria. Los EIM pueden debutar en el periodo neonatal, lactante, infantil o adulto (1-2). El diagnóstico general de los EIM se realiza a través de los siguientes niveles:
1. Identificación de los metabolitos en los fluidos biológicos (orina, sangre, líquido cefalorraquídeo)
2. Cuantificación de la enzima involucrada.
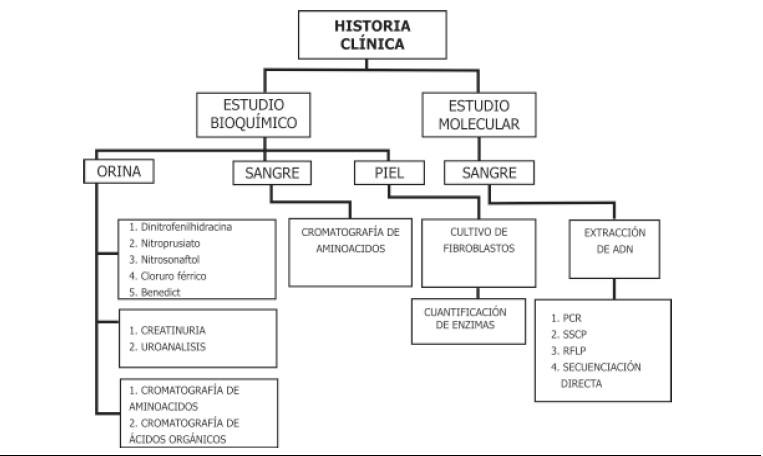
3. Confirmación de la mutación por técnicas de biología molecular. A nivel general el proceso para realizar el diagnostico de los EIM se presenta en la Figura 1.
Figura 1. Diagnóstico de los EIM. Los EIM se estudian siguiendo la secuencia que se presenta en la figura 1. PCR, reacción en cadena de la polimerasa. SSCP. (single strand conformational polymorphism). RFLP, (restriction fragment lenght polymorphism).
Este EIM se caracteriza por el aumento de homocisteína en orina y se debe en la mayoría de los casos a una deficiencia de cistationina â sintasa (EC 4.2.1.22)(CBS). Esta enzima interviene en el catabolismo del aminoácido esencial conocido como metionina Figura 2. La CâS es un homotetrámero dependiente del cofactor piridoxal fosfato (PLP) y condensa la serina con la homocisteína para producir cistationina en un paso irreversible en el proceso de transulfuración de la metionina (3).
Las manifestaciones clínicas de la deficiencia de la CßS son variables con compromiso de diferentes sistemas del organismo como ocular (miopía grave y luxación del cristalino), esquelético (osteoporosis, escoliosis, hábitos marfanoides), sistema vascular (arteriosclerosis, prematura) y sistema nervioso central (retardo mental, convulsiones y trastornos psiquiá- tricos) (4).
Deficiencia de la Adenilosuccinato Liasa (ADSL): Es una enfermedad autosómica recesiva debido a un defecto en la vía de síntesis de las purinas (5), la adenilosuccinato liasa es la enzima que cataliza dos pasos importantes en la vía de formación delATP, el primer paso es la formación de AICAR (aminoimidazol carboxamida ribosil-5-fosfato) a partir de SAICAR (aminoimidazol succinil carboxamida ribosil-5-fosfato), en la síntesis de novo de los nucleótidos de purina. La segunda reacción que cataliza la enzima es la conversión de Adenilsuccinato (S-Ado) a AMP en la ruta donde se forma ATP a partir de IMP, Figura 3. El diagnóstico se realiza con la sospecha clínica seguido de la identificación de succinil purinas SAICAR y S-Ado en líquidos corporales por medio del test de Bratton Marshall en orina (6).
Esta enfermedad cursa con un cuadro clínico muy variable, el principal síntoma en la mayoría de los pacientes diagnosticados es el retardo del desarrollo psicomotor severo (7). En el periodo neonatal se puede encontrar retardo en el crecimiento, hipotonía, movimientos involuntarios de las extremidades, daño o atrofia muscular, espasmo e hipoplasia cerebral, en la infancia el paciente puede cursar con retardo mental, acompañado de convulsiones, epilepsias y rasgos autistas. El gen de la ADSL se encuentra localizado en la región 22q13.1- q13.2 (8)
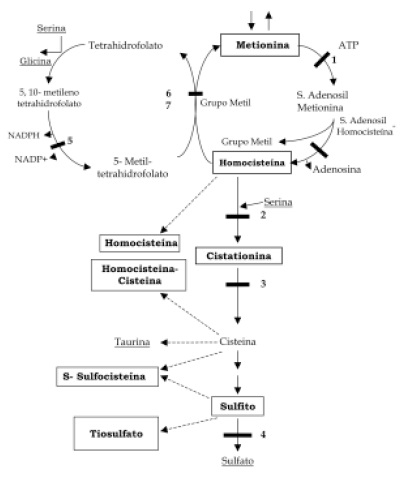
Figura 2. Metabolismo de la homocisteína. La homocisteína es metabolizada por la vía de transulfuración con las enzimas Cistationina â sintasa CBS (2), Cistationasa (3), Sulfito oxidasa (4), y por la vía de la remetilación con las enzimas Metiléntetrahidrofolato reductasa, MTHFR (5), Metionina sintasa (6) y de Betaína –homocisteína metiltransferasa (7). Modificado de Skovby (1996) Tomado de las guías de laboratorio de “Errores Innatos”
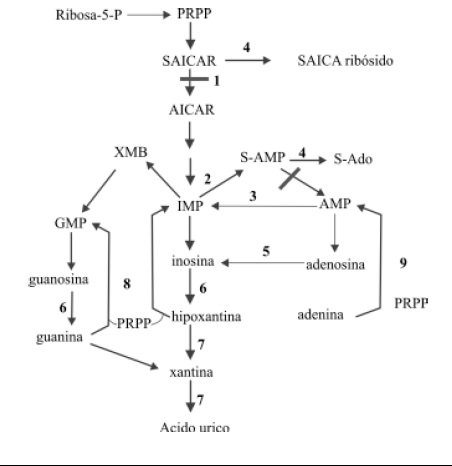
Figura 3. Vía de síntesis de las purinas. Tomado de Yanes AV, Monaga MC (9).
En el presente trabajo se describe la aproximación al diagnóstico de homocistinuria, la confirmación del EIM se realizó cuantificando la actividad de la CBS en fibroblastos y se utilizaron técnicas de biología molecular para detectar la mutación que produce la enfermedad en los pacientes (10). El diagnostico de ADSL se realizó por métodos bioquímicos.
Paciente N°1: sexo masculino, 7 años de edad, normal al nacimiento posteriormente inicia cuadro de convulsiones, retraso psicomotor, problemas oculares y deformidades esqueléticas, características que hicieron sospechar homocistinuria.
Paciente Nº 2: sexo masculino, padres consanguíneos presentó en la época neonatal síntomas de hipotonía, rechazo a la alimentación, antecedente de hermano fallecido en la época neonatal.
La aproximación al diagnóstico de homocistinuria se realizó utilizando la siguiente secuencia:
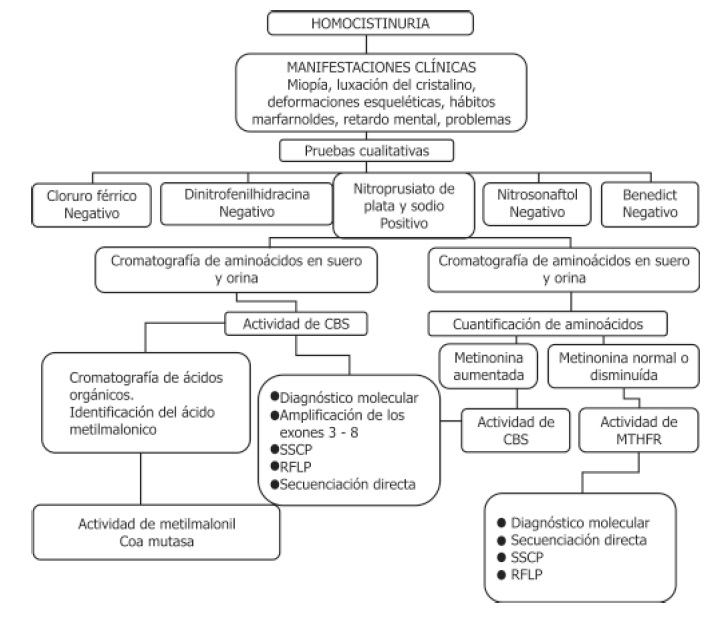
Figura 4. Diagrama diagnóstico para la homocistinuria
La presencia de aminoácidos sulfurados como la homocisteína y cistina son evidenciados con la técnica colorimétrica de nitroprusiato de sodio (11). Utilizando esta técnica los sulfhídrilos oxidados, son reducidos inicialmente con cianuro de sodio, para luego reaccionar con el nitroprusiato de sodio (con grupos sulfhídrilos libres), produciendo una coloración de rosa púrpura. Para diferenciar entre homocisteína y cistina se utiliza la técnica del nitroprusiato de plata: la homocistina es reducida a la forma reactiva (homocisteína) por el nitrato de plata, mientras que la cistina permanece en la forma oxidada (no reactiva). La homocistina reacciona con el nitroprusiato para dar un complejo púrpura.
Posteriormente se realizó cromatografía de aminoácidos en capa fina para separar los aminoácidos en sangre y orina. Esta prueba permitió evidenciar el aumento de metionina en sangre y en orina. Luego se realizó la cuantificación de estos aminoácidos utilizando cromatografía liquida de alta resolución, y el método inmunológico IMX de Abbott© para cuantificar la homocisteína plasmática. Se realizó cromatografía de gases acoplado a espectrofotometría de masas para descartar un aumento de ácido metilmalónico y de homocisteína, como consecuencia de la deficiencia de vitamina B12. La cuantificación de la enzima CßS se realizó en cultivo de fibroblastos, procedentes de biopsia de piel, utilizando como sustratos la homocistina y la serina marcada con 14C. Para aplicar técnicas de biología molecular fue necesario hacer extracción de DNA, por el método de Wizard. Aplicando la técnica de la reacción en cadena de la polimerasa (PCR), posteriormente se procedió a realizar se amplificaron los exones 3 y 8 de la CßS, donde se han reportado el mayor número de mutaciones (3).
. SSCP (single strand conformational polymorphism): se utiliza para delimitar el lugar de la mutación en un exón específico de un gen determinado. Consiste en desnaturalizar los fragmentos de DNA amplificados y separarlos en un gel de acrilamida en condiciones no desnaturalizantes. El DNA durante la electroforesis va adquiriendo estructuras complejas dependiendo de su conformación que viene dictaminada por la secuencia de nucleótidos, el cambio de una sola base es suficiente para producir un patrón de bandas diferentes de los que no tiene la mutación (11).
RFLP (restriction fragment lenght polymorphism): Se utilizan enzimas que van a reconocer una secuencia especifica de nucleótidos (sitio de restricción) en el producto amplificado del ADN; usualmente son de 4 a 10 pb denominándose diana de restricción y cortan el ADN en esa zona (11)
Cromatografía liquida de alta resolución en fase reversa: El sistema HPLC esta acoplado a un detector de diodos que permite al análisis espectral a 254 a 280 nm. La identificación de succinil purinas se realiza por el método descrito por Simmonds (11) en orina, y modificado para plasma y liquido cefalorraquídeo (LCR) así: 50 ml del volumen del ultrafiltrado de LCR o plasma. El gradiente lineal se correrá con el solventeAal 100 % (acetato de amonio, ajustado a un pH 5 con ácido acético glacial) y el solvente B al 20 % (metanol-acetonitritotetrahidrofurano 80:10:10, v/v) por más de 40 minutos usando un índice de flujo de 0.5 ml/min.
La deficiencia de adenilosuccinato liasa se diagnostic ó según el esquema que aparece al inicio de la página siguiente.
. Test de Bratton-Marshall: Esta prueba detecta SAICAR, mediante la amina libre en el compuesto de SAICAR reacciona con el nitrito de sodio formándose una sal en un medio ácido, luego, este compuesto forma un complejo con N-1-napthylenediamine
dihydrochloride (NEDA) dando como resultado una coloración púrpura. Posteriormente se realiza una curva de calibración para determinar las concentraciones de SAICAR en orina y otros líquidos corporales teniendo en cuenta que la absorción del compuesto se lee a 555 nm con un patrón comercial (6).
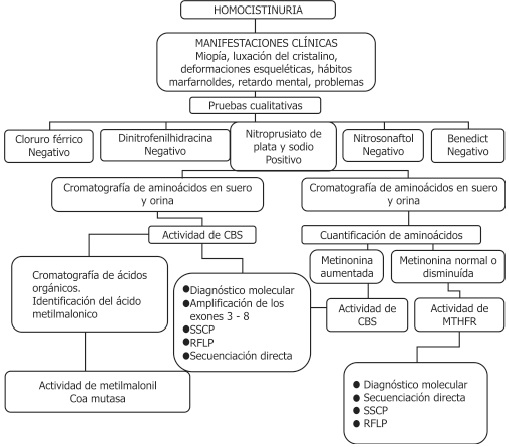
Figura 5. Diagrama diagnóstico para la deficiencia de adenilosuccinato liasa
Tabla 1. Identificación y cuantificación de metabolitos de metionina en sangre, orina en el paciente N°1.
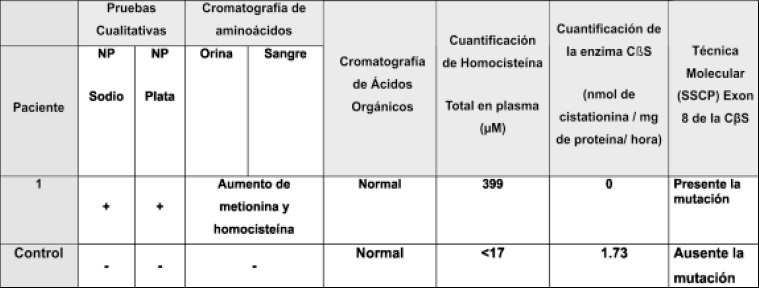
En la tabla 1 se presentan los resultados correspondientes a los análisis cualitativos (nitroprusiato de sodio y de plata) y cuantitativos (cuantificación de homocisteína y cuantificación de la actividad de CßS en cultivo de fibroblastos. Identificación de una mutación en el exon 8 de la CßS así como la identificación de la mutación que afecta al paciente N° 1 con características clínicas de homocistinuria.
El tamizaje molecular para identificar la mutación utilizando SSCP mostró los resultados que se muestran en la Figura 6.
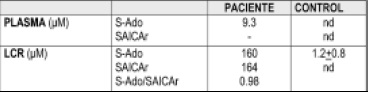
En el paciente N° 2 los resultados fueron los siguientes: la prueba cualitativa o test de Bratton-Marshall en muestra de orina dio color púrpura el cual evidencia la presencia de metabolitos de las purinas. En la anterior tabla se muestran los metabolitos de las purinas (S-Ado, SAICAR, S-Ado/ SAICAR) que se encontraron aumentadas en el paciente N° 2
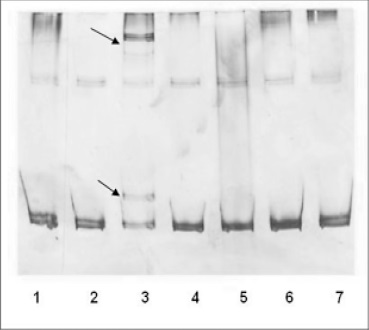
Figura 6. SSCP del exón 8 de cistationina ß sintasa. Carril 1: control normal; Carriles 2, 4, 5, 6, 7: individuos afectados con patrón de migración normal; Carril 3: paciente N°1 afectado con patrón de migración anormal.

Figura 7. Test De Bratton-Marshall.
Tabla 2. Análisis de succinilpurinas en plasma y líquido cefalorraquídeo del paciente N° 2 LCR: Líquido Cefalorraquídeo, S-Ado; succiniladenosina; SAICAr: succinilaminoimidazole carboxamida ribosa;nd: no detectable.
Los EIM son trastornos bioquímicos de origen genético. El diagnostico temprano permite instaurar el tratamiento para evitar las secuelas que estos defectos producen. En el caso de no disponer de un tratamiento efectivo, es importante hacer el diagnóstico para realizar consejería genética a la familia. Entre estas enfermedades se encuentra la homocistinuria y la deficiencia de adenilosuccinato liasa las cuales producen desequilibrios químicos importantes en el organismo provocando un deterioro gradual de la salud.
Para realizar un diagnóstico preciso de estas enfermedades se debe tener en cuenta la presentación clínica, la cual a pesar de ser variable, muestra algunas manifestaciones clínicas que son orientadoras. En la homocistinuria las características clínicas más importantes están la aparición de problemas oculares antes de los 5 años y deformidades esqueléticas antes de los 15 años. En el caso de ADSL la presencia de severa hipotonía en el periodo neonatal y en la infancia retardo mental, convulsiones, epilepsias y rasgos autistas son manifestaciones clínicas de este defecto metabólico.
Una vez establecida la sintomatología del paciente se procede a realizar las pruebas químicas que dan una orientación del problema metabólico; en la homocistinuria el nitroprusiato de sodio y de plata son positivos. El aumento de metionina y homocisteína encontrado al separar y cuantificar estos metabolitos permiten establecer con mayor certeza el diagnóstico que es confirmado por cuantificación de la enzima involucrada (14). La búsqueda de la mutación se puede abordar utilizando SSCP Figura 6, que mostrará un patrón de bandas diferentes entre los normales y los que tienen la mutación, a estos últimos se realiza secuenciación del exón correspondiente y la confirmación de la mutación mediante RFLP.
Es importante verificar si la mutación encontrada responde al tratamiento que consiste en la combinación de piridoxina, ácido fólico y vitamina B12, de lo contrario es necesario hacer restricción de metionina y suplemento de cistina y adicionar betaína para la remetilación de la homocisteína a metionina (3). Realizar una secuencia de estudio utilizando métodos químicos, bioquímicos y moleculares como los que se describen en este trabajo, permitirán identificar y confirmar el defecto metabólico que conduce a las manifestaciones clínicas de la enfermedad, como en el caso de la homocistinuria. Por otra parte, establecer la respuesta al tratamiento y hacer un pronóstico del resultado del tratamiento.
Para la deficiencia de ADSL, el diagnóstico se basa en la detección de metabolitos marcadores presentes en los fluidos biológicos orina sangre y líquido cefalorraquídeo, este aumento implica que el metabolismo de las purinas está alterado y como consecuencia la síntesis de macromoléculas que incorporan estas bases nitrogenadas estará afectado.
La identificación de esta enfermedad metabólica se realiza fácilmente utilizando el test de Bratton- Marshall Figura 7, método es simple, de bajo costo, que utiliza muestras de orina impregnadas en papel de filtro facilitando, el transporte y el almacenamiento de las muestras.
Hasta el momento no está disponible un tratamiento efectivo, sin embargo, algunos pacientes han sido tratados con administración oral de D-ribosa. Se recomienda que en todos los pacientes con afectación neurológica de etiología desconocida sea investigado este desorden metabólico con el fin de proporcionar consejería genética a los padres (14).
. Las pruebas colorimétricas (nitroprusiato de sodio, nitroprusiato de plata y el test de Bratton-Marshall) son sencillas, de bajo costo y muy útiles en la aproximación al diagnóstico de los EIM como es el caso de la homocistinuria y la deficiencia de ADSL respectivamente.
. Las técnicas moleculares permiten detectar la mutación que conducen a las manifestaciones clínicas de la enfermedad y conocer la respuesta al tratamiento favoreciendo de esta manera un manejo adecuado para el paciente.
1. Bermúdez M. Trastornos bioquímicos de origen genético. Sistema de información interactivo. III semestre de Medicina. Guías de errores innatos del metabolismo. Instituto de genética humana. Universidad Pontificia Javeriana. 2005
2. Sanjurjo Crespo P, Aquino L, Aldámiz E. Enfermedades cong énitas del metabolismo: generalidades, grupos clínicos y algoritmos diagnósticos. En: Diagnostico y tratamiento de las enfermedades metabólicas hereditarias. Sanjurjo P, Baldellou A. Madrid. Ediciones Ergon S. A, 2001. 1-13.
3. Mudd DSH, Levy HL, Kraus JP. Disorders of transsulphuration. In: The metabolic and molecular bases of inherited disease. Scriver, Beaudet, Sly, Valle, editors.8a. Edition. New York: McGraw Hill; 1995. 56. p. 2001-2007
4. Gaustadnes M, Wilcken B, Oliveriusova J, McGill J, Fletcher J. The molecular basis of cystathionine â synthase deficiency in Australian patients; genotype- phenotype correlations and response to treatment. Human Mutat. 2002; 20: 117-26
5. Castro M, Pérez C, Merinero B, García MJ, Bernal J, Gil ángel A, Bermúdez M., Screening for adenylosuccinate lyase deficiency: clinical, biochemical and molecular findings in tour patients. J Pediatr Neurob, Neurol and Neurog. 2002; 33: 186-189.
6. Laikind P.K, Seegmiller J.E, Gruber H. E. Detection of 5’- p h o s p h o r i b o s y l - 4 - ( N - s u c c i n y l c a r b o x a m i d e ) - 5 - aminoimidazole in orine by use of the Bratton-Marshall reaction: identification of patients deficient in 55 Adenylsuccinate lyase activity. Analytical biochemistry. 1986; 156: 81-90.
7. Van den Bergue G, Vincent MF, Jaeken J. Inborn errors of the purine nucleotide cycle: Adenylosuccinase deficiency. J Inher Metab Dis.1997; 20: 193-202.
8. Van Keuren ML, Hart IM, Kao FT, Neve RL, Brauns GAP, Kurnit DM et al. A somatic cell hybrid with a single human chromosome 22 corrects the defect in the CHO mutant (Ade- 1) lacking Adenylosuccinate activity. Cytogenet Cell Genet.1987; 44:142-7.
9. Yanes AV, Monaga MC. Deficiencia de adenilosuccinato liasa: un breve repaso. Biomédica. 2004; 15:243-250.
10. Desviat LR, Perez B, Ugarte M. Bases moleculares de las enfermedades metabólicas hereditarias. En diagnóstico y tratamiento de las enfermedades metabólicas hereditarias. Sanjurjo P, Baldellou A. Ed. Madrid. Ediciones Ergon S.A, 2001. 1-13p.
11. Thomas G, Rodney H. Selected screening test for genetic metabolic disease: Yearbook Medical Publishers, Inc. 1973.
12. Simmonds HA, Duley JA, Davies PM. Analysis of purines and pyrimidines in blood, urine and other physiological fluids. In: Hommes F editor. Techniques in diagnostic human biochemical genetics: a Laboratory manual. New York: Wiley-Liss; 1991. 397-424 p.
13. Bermúdez M, Bernal J, Espinosa E, Cornejo W, Briceño I, Prieto JC et al. Propuesta para un protocolo de diagnóstico bioquímico de homocistinuria. Universitas Medica. 2003; 44 (3): 119-124
14. Sivendran S, Patterson D, Spiegel E, McGown I, Cowley D, Colman R. Clinical biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Human Mol Genet. 2000; 9 (14): 2159-2165.