Carolina Andrea Infante Molina 1, 2, Bacterióloga; Laura Marlen Mora Forero1, 2, Bacterióloga; Jenny C. Ortega Rojas 1, M. Sc; Carlos E. Arboleda-Bustos1, Ph.D; William Fernández1, Ph.D; Humberto Arboleda1, M. Sc; Gonzalo Arboleda1, Ph.D.
1. Grupo de Neurociencias, Facultad de Medicina e Instituto de Genética, Universidad Nacional de Colombia.
2. Universidad Colegio Mayor de Cundinamarca. Bogotá, Colombia.
Correspondencia: gharboledab@unal.edu.co
Recibido: 25/02/2014 Aceptado:05/06/2014
La enfermedad de Parkinson es un desorden neurodegenerativo complejo, caracterizado por la pérdida progresiva de las neuronas dopaminérgicas de la sustancia nigra pars compacta. Fac-tores tanto ambientales como genéticos se ha determinado que contribuyen a su desarrollo. Mutaciones en los genes PINK1 y PARKIN han sido asociadas con la enfermedad de inicio temprano e historia familiar. El objetivo del presente estudio fue identificar mutaciones en los genes PINK1 (exones 4 y 6) y PARKIN (exones 2 y 7) en 22 pacientes colombianos con EP de inicio temprano y/o antecedentes familiares, mediante amplificación por PCR y secuenciamiento. Las secuencias se compararon con la secuencia consenso de referencia. Se detectó una mutación homocigota de cambio en el marco de lectura ( frameshift) c.155delA en el exón 2 del gen PARKIN en una paciente con inicio temprano de la enfermedad e historia familiar. Además se identificó la presencia de un polimorfismo en el intrón 2 del gen PARKIN en siete pacientes, uno de ellos en estado homocigoto. No se encontraron mutaciones en los exones 4 y 6 del gen PINK1. Se encontró una mutación homocigota c.155delA en el exón 2 de PARKIN de una paciente con la enfermedad de Parkinson de inicio temprano con historia familiar. No se encontraron cambios el gen PINK1.
Palabras clave: enfermedad de Parkinson, mutaciones, PCR, secuenciación directa de ADN.
Parkinson's disease is a complex neurodegenerative disorder, characterized by the progressive loss of dopaminergic neurons of the substance nigra pars compacta. It has been determined that factors both environmental and genetic contribute to its development. Mutations in the genes PINK1 and PARKIN have been associated with the early onset of disease and family history. The goal of this study was to identify mutations in the PINK1 genes (exons 4 and 6) and PARKIN (exons 2 and 7) in 22 Colombian patients with EP of early onset and/or family history, by PCR amplification and sequencing. The sequences were compared with the reference consensus sequence. A homozygous change mutation was detected in the reading frame (frame shift) c.155dela in exon 2 of the PAR-KIN gene in a patient with early onset of the disease and family history. In addition, the presence of a polymorphism in intron 2 of the PARKIN gene was identified in seven patients, one of them in homozygous state. Mutations were not found in exons 4 and 6 of the gene PINK1. A homozygous mutation c.155dela in exon 2 of PARKIN was found in a female patient with Parkinson's disease early onset with family history. No changes to the gene PINK1 were found.
Key Words: direct DNA sequencing, mutations, Parkinson's disease, PCR
La enfermedad de Parkinson (EP) es el segundo desorden neurodegenerativo más común después de la enfermedad de Alzheimer (1, 2), caracteriza-do por la pérdida progresiva de las neuronas do-paminérgicas de la sustancia nigra pars compac-ta (SNpc) y la presencia de cuerpos de Lewy en las neuronas que sobreviven a esta muerte celular (3). Según la Organización Mundial de la Salud (OMS) la EP afecta aproximadamente a 6.3 mi-llones de personas en todo el mundo. En Colom-bia en el 2003 se reportó una prevalencia de EP de 4.7 por 1000 habitantes (4) y en 2011 se alcan-zaron cifras de 230 mil personas afectadas, según la Liga colombiana de Parkinson.
La causa de la EP no se conoce en su totalidad, pero la interacción de factores ambientales y genéticos se asocian a su etiología, lo que determina la pre-sentación de la enfermedad en su forma esporádica (90-95%) o familiar (5-10%) (2,5). Para el 2012, 18 locus han sido asociados con la enfermedad (6). PINK1 Y PARKIN son genes cuyas mutaciones hansido asociadas a la EP de inicio temprano (EOPD: del inglés Early-OnsetPakinson´s Disease), tanto en la formas familiares como esporádicas (7-9). En la presentación familiar las mutaciones en estos genes se heredan con un patrón autosómico recesivo. La prevalencia de las mutaciones en estos dos genes varía dependiendo del grupo étnico y la zona geográfica (7). Para PINK1 se reporta una prevalencia de 1-9% en los casos de inicio tem-prano (2,10,11) y para PARKIN en un 50% en casos de inicio juvenil, 10-25% en casos espo-rádicos de inicio temprano y menos del 5% en casos esporádicos de inicio tardío (2,11,12). En Colombia son pocos los estudios acerca de mu-taciones en estos genes.
PINK1 codifica para una serina treonina quina-sa (10,13) y PARKIN codifica para una ligasa de ubiquitina E3, que participa en mecanismos de degradación proteica (14,15). Las funciones de estas proteínas en la EP no se han esclarecido por completo, pero de forma importante par-ticipan en vías de regulación de la homeostasis mitocondrial y procesos de mitofagia (2,13,16). El propósito del presente estudio fue identificar mutaciones en los genes PINK1 (exones 4 y 6) y PARKIN (exones 2 y 7) en pacientes colombia-nos con EP de inicio temprano y/o antecedentes familiares, mediante amplificación por PCR y secuenciamiento.
Pacientes
Se analizaron un total de 22 muestras de pacientes colombianos con EP de inicio temprano y/o historia familiar escogidos por el método de muestreo no probabilístico intencional o por conveniencia aplicando los criterios de inclusión: (i) inicio temprano y/o historia familiar, (ii) Los pacientes fue-ron valorados previamente por un grupo de profesionales del grupo de Movimientos Anormales de la Universidad Nacional de Colombia, mediante los criterios de la Parkinson’s UK Brain Bank (17-19). La edad de inicio temprano de la enfermedad se tomó como presencia de signos de Parkinson antes de los 40 años (7, 20); inicio juvenil antes de los 20 años (21, 22). Se obtuvo una muestra de sangre periférica por venopunción, con firma previa del consentimiento informado.
Extracción de ADN genómico
Para la extracción de ADN genómico se utilizó el kit comercial, Quick-gDNA TM MiniPrep (ZYMO Research Corp, Irvine, Estados Unidos), siguiendo el protocolo establecido. La comprobación de la extracción se realizó mediante electroforesis en gel de agarosa al 0.8%.
Amplificación por PCR
La amplificación de PINK1 (exones 4 y 6) y PARKIN (exones 2 y 7) se realizó por medio dela reacción en cadena de la polimerasa (PCR) utilizando primers específicos previamente re-portados (21, 23) que amplifican las regiones de unión intrón-exón para las regiones de in-terés. Las concentraciones finales de los reactivos para la amplificación de todos los exones fueron las mismas: buffer 1X, 0.4μM de cada uno de los primers, 10mM de dNTPS y 0.625U de taq polimerasa; para los exones 6 de PINK1 y 2 de PARKIN la concentración de MgCl 2 fue de 1mM y para los exones 4 de PINK1 y 7 de PARKIN fue de 1.5mM.
El programa de PCR utilizado para los exones de PINK1 y el exón 2 de PARKIN fue de una etapa de denaturación inicial a 95°c por tres minutos, 1 ciclo de denaturación a 95°c por 30 segundos, hibridación (exón 4: 63.2°C; exón 6: 56.9°C; y exón 2: 55.0°C, todos por 30 segun-dos) y extensión a 72°C, por 30 ciclos; y una extensión final a 72°C por 5 minutos. Para el exón 7 de PARKIN se utilizó una PCR con un protocolo de rampa decreciente de temperaturas que fueron bajando de 58°a 48°C, es decir 0.5°C por ciclo.
Secuenciación directa de ADN
Para la purificación de los amplificados se uti-lizó el método de etanol-acetato de amonio y finalmente se realizó una mezcla de secuencia-ción para su procesamiento en el servicio de Secuenciación del Instituto de Genética de la Universidad Nacional de Colombia mediante la metodología de BigDye siguiendo el protocolo del fabricante (Applied Biosystem, Foster City, Estados Unidos). Para el análisis de los cromato-gramas se utilizaron los programas bioinformáti-cos Bioedit (24) y Chromas (Technelysium Pty Ltd.) para determinar la calidad de las secuencias y NovoSnp (25) para realizar la alineación de las secuencias con el gen de referencia.
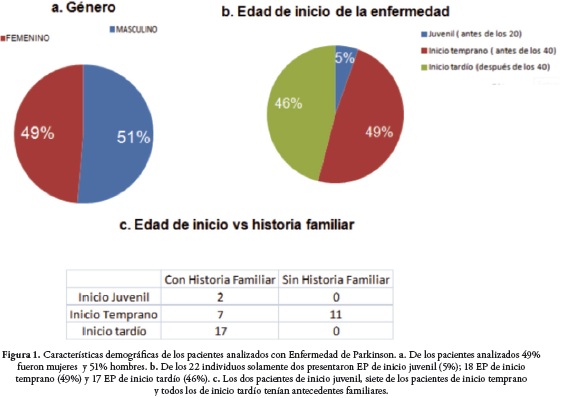
De los 22 pacientes con EP de inicio temprano y/o historia familiar analizados, 18 fueron mujeres y 19 hombres, Figura 1.
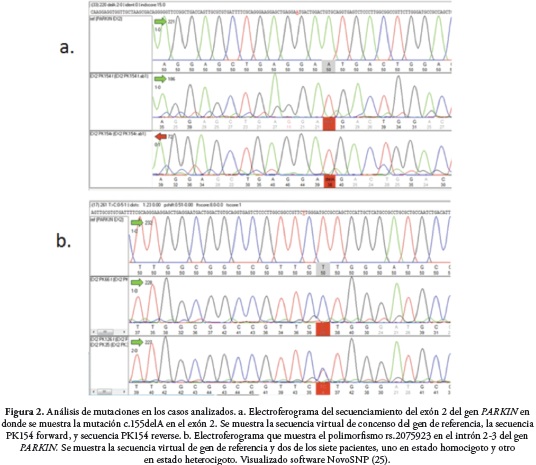
Después de analizar las 88 secuencias, se encontró en la muestra PK154 una mutación de cambio en el marco de lectura (frameshift) c.155delA en el exón 2 del gen PARKIN (Figura 2a), la cuál ha sido reportada en otras poblaciones incluyendo la colombiana (26-28). En los 21 pacientes restantes no se encontraron otras alteraciones en el gen PARKIN.
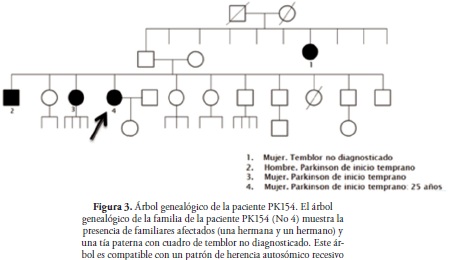
La paciente PK154 es proveniente de Santa Lucía, Nariño (Colombia). La historia familiar refiere un hermano y una hermana afectados con EOPD, Figura 3. Su cuadro clínico inició a los 25 años con dificultad para caminar, dada por rigidez y pérdida del equilibrio, seguido por debilidad de miembros inferiores y bradiquinesia, con posterior compromiso de miembro superior izquierdo y luego el derecho, acompañado de temblor y rigidez marcada. Posteriormente se asoció a rigidez en articulación temporomandibular. La paciente recibió tratamiento con levodopa con una adecuada respuesta pero posteriormente presentó disquinesias secundarias, por lo que se le suspendió. Por otro lado, la paciente manifestó signos no motores de depresión.
Se encontró además un polimorfismo rs2075923 en siete pacientes, que corresponde al cambio de una timina por un citosina en el intrón 2-3 de PARKIN, uno en estado homocigoto, el cuál hasido previamente reportado (26,29,30), Figura 2b. Por otra parte, no se encontraron alteraciones en el gen PINK1 en la muestra analizada.
En este estudio se realizó un análisis mutacional en los genes PINK1 (exones 4 y 6) y PARKIN (exones 2 y 7). Tras el análisis de las 88 secuencias se identifi-có una mutación homocigota c.155delA en el exón 2 del gen PARKIN en una paciente de inicio tempra-no a los 25 años e historia familiar de EP, mutación que ha sido previamente en otros reportes incluyendo uno en población colombiana (28,31,32). El fenotipo asociado con mutaciones en PARKIN es clínicamente indistinguible de la EP idiopática, pero usualmente se caracteriza por inicio temprano alrededor de los 30 años y progresión lenta, como en el caso de la paciente analizada en este estudio. Además presentó disquinesias inducidas por levodopa tiempo después del tratamiento, que es también frecuente en pacientes con mutaciones en PARKIN (2, 31).
La mutación c.155delA produce una alteración del marco de lectura (frameshift) cambiando el codón AAT por ATG que conlleva al cambio de una asparagina por una metionina en la posición 52 de la proteína. Además genera un codón de parada prematuro en la posición 81, produciendo una proteína aberrante y truncada (26,32,33) la cual pierde 385 aminoácidos presentes en Parkin (28). Esta mutación se hereda en una forma autosómica recesiva generan-do una pérdida de función, ya que afecta el domino UBL en el extremo N-terminal, importante para su función como ligasa E3 de ubiquitinas, reconociendo proteínas blanco de degradación (26,34).
Parkin es una ligasa E3 de ubiquitinas que junto con Pink1 participa en una vía común para el mantenimiento de la homeostasis mitocondrial y mitofagia (13,16). En caso de una alteración como la mutación c.155delA, donde PARKIN pierde su actividad enzimática, se ven alterados procesos de remoción óptima de mitocondrias dañadas, por la falta de ubiquitinación requerida para este proceso, ocasionando la liberación de mediadores proapoptóticos que culminan en el proceso de muerte neuronal (16,35). Adicionalmente, la pérdida de PARKIN incide en la acumulación de proteínas mal plegadas y agregados proteicos que no son adecuadamente de-gradados por el sistema de ubiquitina-proteosoma, los cuales también contribuyen al proceso neurodegenerativo (36,37). Todo lo anterior conlleva a una pérdida de las neuronas dopaminergicas en la sustancia nigra pars compacta, con disminución de los niveles de dopamina lo que se ve reflejado en el cuadro clínico de los pacientes con EP.
Para conocer el efecto de esta mutación en procesos celulares como la regulación de la transcripción y traducción de la proteína se hacen necesarios estudios de epigenética y de RNAm; además de estudios funcionales en diferentes tipos celulares para evaluar dinámica mitocondrial, mecanismos de degradación proteica y muerte celular.
En Colombia, la presencia de esta mutación ya se ha reportado previamente, encontrándose en forma homocigota en una familia consanguínea (28). Dado el gran número de reportes de esta mutación en Europa, especialmente en Francia y España (26,27,38), se ha sugerido que la mutación tendría un origen ancestral europeo asociado a la colonización y mezcla poblacional en Colombia (28). Según el análisis genealógico de la paciente PK154, se podría inferir que sus dos hermanos afectados, con inicio temprano de la enfermedad, son homocigotos para la misma mutación. Así mismo, se puede deducir que sus padres y su hija son portadores heterocigotos aparentemente sanos. Al parecer el estado heterocigoto inferido en los padres e hija no conlleva al desarrollo de la enfermedad, como ha sido reportado en estudios previos en los que se ha encontrado la mutación en estado heterocigoto en individuos asintomáticos (27,31,39). Sin embargo, otros estudios han encontrado esta mutación en estado heterocigoto en paciente con EP (7,40) y casos de heterocigotos compuestos (41). De tal forma que el papel fisiopatológico de esta mutación en forma heterocigota sigue siendo controversial (7,40). Se sugiere evaluar la presencia de esta mutación en estos familiares y si es posible en otros familiares.
No se encontraron cambios en las secuencias analizadas del gen PINK1, similar a lo descrito en otras poblaciones (1,42,43). El polimorfismo observado (IVS2 + 25T>C; rs2075923) ha sido previamente reportado (26,30) y no se ha encontrado una asociación entre esta variante y la EP (29).
Los resultados de esta investigación corroboran la baja frecuencia de mutaciones en los genes PINK1 y PARKIN en las regiones analizadas en casos con historia familiar o inicio temprano de la EP en población colombiana. Es necesario, realizar un estudio a mayor escala que vincule la totalidad del gen y análisis de variaciones en el número de copias de estos genes. Además, es importante la realización de estudios de asociación genética de variantes en estos dos genes para definir su participación como factores de riesgo en la EP esporádica.
Este trabajo fue financiado con recursos del proyecto de la DIB-Universidad Nacional de Colombia y Colciencias.
1. Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Ge-netic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat. 2010;31(7):763-780.
2. Qayyum A. Etiology and Pathophysiology of Parkinson’s Di-sease. In: Rana AQ, ed: InTech; 2011.
3. Fahn S, Jankovic J, Hallett M. Chapter 5 - Current concepts on the etiology and pathogenesis of Parkinson disease. Princi-ples and Practice of Movement Disorders 2° Edition ed. Edin-burgh: W.B. Saunders; 2011:93-118.
4. Pradilla AG, Vesga AB, Leon-Sarmiento FE. [National neu-roepidemiological study in Colombia (EPINEURO)]. Rev Panam Salud Publica. 2003;14(2):104-111.
5. Pankratz ND, Wojcieszek J, Foroud T. Parkinson Disease Overview. 2009.
6. Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous muta-tions in genes associated with parkinsonism. Lancet Neurol. 2007;6(7):652-662.
7. Camargos ST, Dornas LO, Momeni P, Lees A, Hardy J, Single-ton A, et al. Familial Parkinsonism and early onset Parkinson’s disease in a Brazilian movement disorders clinic: phenotypic characterization and frequency of SNCA, PRKN, PINK1, and LRRK2 mutations. Mov Disord. 2009;24(5):662-666.
8. Scornaienchi V, Civitelli D, De Marco EV, Annesi G, Tarantino P, Rocca FE, et al. Mutation analysis of the PINK1 gene in Southern Italian patients with early- and late-onset parkinso-nism. Parkinsonism Relat Disord. 2012;18(5):651-653.
9. Yonova-Doing E, Atadzhanov M, Quadri M, Kelly P, Shawa N, Musonda ST, et al. Analysis of LRRK2, SNCA, Parkin, PINK1, and DJ-1 in Zambian patients with Parkinson’s disea-se. Parkinsonism Relat Disord. 2012;18(5):567-571.
10. Thomas B, Beal MF. Parkinsons disease. Hum Mol Genet. 2007;16 Spec No. 2:R183-194.
11. Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol. 2011;26 Suppl 1:S1-58.
12. Fung HC, Chen CM, Hardy J, Singleton AB, Lee-Chen GJ, Wu YR. Analysis of the PINK1 gene in a cohort of patients with sporadic early-onset parkinsonism in Taiwan. Neurosci Lett. 2006;394(1):33-36.
13. Cookson MR, Bandmann O. Parkinson’s disease: insights from pathways. Hum Mol Genet. 2010;19(R1):R21-27.
14. Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Mino-shima S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25(3):302-305.
15. Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000;97(24):13354-13359.
16. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochon-dria to activate Parkin. PLoS Biol. 2010;8(1):e1000298.
17. Douglas J. Diagnostic criteria for Parkinson disease. . Archives of Neurology. 1999; Vol 56(1): 33-39.
18. Clarke CE. Parkinson’s disease. Bmj. 2007;335(7617):441-445.
19. Jankovic J. Parkinsons disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368-376.
20. Sironi F, Primignani P, Zini M, Tunesi S, Ruffmann C, Ricca S, et al. Parkin analysis in early onset Parkinson’s disease. Par-kinsonism Relat Disord. 2008;14(4):326-333.
21. Kitada T, Asakawa S, Hattori N, Matsumine H, Yama-mura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605-608.
22. Periquet M, Latouche M, Lohmann E, Rawal N, De Michele G, Ricard S, et al. Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain. 2003;126(Pt 6):1271-1278.
23. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158-1160.
24. Hall TA. BioEdit: a user-friendly biological sequence align-ment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95-98.
25. Weckx S, Del-Favero J, Rademakers R, Claes L, Cruts M, De Jonghe P, et al. novoSNP, a novel computational tool for sequen-ce variation discovery. Genome Res. 2005;15(3):436-442.
26. Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De Mi-chele G, et al. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Euro-pe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum Mol Genet. 1999;8(4):567-574.
27. Munoz E, Tolosa E, Pastor P, Marti MJ, Valldeoriola F, Cam-pdelacreu J, et al. Relative high frequency of the c.255delA parkin gene mutation in Spanish patients with autosomal recessive parkinsonism. J Neurol Neurosurg Psychiatry. 2002;73(5):582-584.
28. Pineda-Trujillo N, Dulcey Cepeda A, Arias Pérez W, More-no Masmela S, Saldarriaga Henao A, Sepúlveda Falla D, et al. Una mutación en el gen PARK2 causa enfermedad de Parkinson juvenil en una extensa familia colombiana. Iatreia. 2009;22 (2):122-131.
29. Oliveira SA, Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, et al. Association study of Parkin gene poly-morphisms with idiopathic Parkinson disease. Arch Neurol. 2003;60(7):975-980.
30. Solla P, Cannas A, Floris G, Murru MR, Corongiu D, Tranquilli S, et al. Parkin Exon Rearrangements and Sequence Variants in LRRK2 Mutations Carriers: Analysis on a Possible Modifier Effect on LRRK2 Penetrance. Parkinsons Dis. 2010;2010:537698.
31. Hoenicka J, Vidal L, Morales B, Ampuero I, Jimenez-Jimenez FJ, Berciano J, et al. Molecular findings in familial Parkinson disease in Spain. Arch Neurol. 2002;59(6):966-970.
32. ras J, Guerreiro R, Ribeiro M, Morgadinho A, Januario C, Dias M, et al. Analysis of Parkinson disease patients from Por-tugal for mutations in SNCA, PRKN, PINK1 and LRRK2. BMC Neurol. 2008;8:1.
33. Dachsel JC, Mata IF, Ross OA, Taylor JP, Lincoln SJ, Hinkle KM, et al. Digenic parkinsonism: investigation of the synergistic effects of PRKN and LRRK2. Neurosci Lett. 2006;410(2):80-84.
34. Rawal N, Periquet M, Lohmann E, Lucking CB, Teive HA, Ambrosio G, et al. New parkin mutations and atypical phe-notypes in families with autosomal recessive parkinsonism. Neurology. 2003;60(8):1378-1381.
35. Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mi-tochondria. Nat Commun. 2012;3:1016.
36. Tan JM, Wong ES, Lim KL.Protein misfolding and ag-gregation in Parkinson’s disease. Antioxid Redox Signal. 2009;11(9):2119-2134.
37. Johnson BN, Berger AK, Cortese GP, Lavoie MJ. The ubi-quitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc Natl Acad Sci U S A. 2012;109(16):6283-6288.
38. Lesage S, Lohmann E, Tison F, Durif F, Durr A, Brice A. Rare heterozygous parkin variants in French early-onset Parkinson disease patients and controls. J Med Genet. 2008;45(1):43-46.
39. Hedrich K, Marder K, Harris J, Kann M, Lynch T, Meija-Santana H, et al. Evaluation of 50 probands with early-onset Parkinson’s disease for Parkin mutations. Neurology. 2002;58(8):1239-1246.
40. Brooks J, Ding J, Simon-Sanchez J, Paisan-Ruiz C, Sin-gleton AB, Scholz SW. Parkin and PINK1 mutations in early-onset Parkinson’s disease: comprehensive screening in publicly available cases and control. J Med Genet. 2009;46(6):375-381.
41. Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol. 2006;63(6):826-832.
42. |Chung EJ, Ki CS, Lee WY, Kim IS, Kim JY. Clinical features and gene analysis in Korean patients with early-onset Parkin-son disease. Arch Neurol. 2006;63(8):1170-1174.
43. Deng H, Le W, Shahed J, Xie W, Jankovic J. Mutation analysis of the parkin and PINK1 genes in American Cau-casian early-onset Parkinson disease families. Neurosci Lett. 2008;430(1):18-22.