Incorporación química de la sonda fluorescente FlAsH en la proteína Hha: aplicación al estudio del complejo Hha/H-NS
Catalina Granados Acevedo1, Tiago N. Cordeiro2, Miquel Pons2,3
1 Programa de Ingeniería Biomédica. Escuela de Ingeniería de Antioquia–Universidad CES.
2Laboratorio NMR Biomolecular, Parc Cientific de Barcelona.
3Universitat de Barcelona.
Correspondencia: catagranados@gmail.com
Recibido: 18-03-09 / Aceptado: 22-04-09
Palabras clave: anisotropía de fluorescencia, FlAsH, Hha, H-NS, proteínas asociadas al nucleoide, sonda molecular.
Keywords: fluorescente anisotropy, FLAsH, Hha, H-NS, molecular probe, nucleoid-associated proteins.
La proteína H-NS (Histone-like Nucleoid Structuing protein) fue caracterizada por primera vez como proteína asociada al nucleoide bacteriano por Varshavsky y colaboradores en 1977 (2), aunque no fue descrita como tal hasta siete años después (3,4). La proteína H-NS ha sido estudiada principalmente en E. coli y S. enterica, donde interviene en la organización del nucleoide y en la regulación de la expresión de genes involucrados en la adaptación a cambios ambientales (5). Es una proteína termoestable y de pequeño tamaño (aproximadamente 15,6 kDa y 137 aminoácidos) (6), pero muy abundante (20.000 copias por célula) (7). Los niveles de expresión de la proteína H-NS se mantienen relativamente constantes a lo largo de las distintas fases de crecimiento (4).
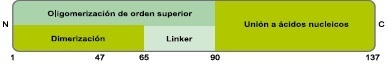
La proteína H-NS presenta tres componentes estructurales, Figura 1. Un dominio N-Terminal, que se extiende desde el residuo aminoacídico 1 hasta el 65, un dominio C-Terminal que se extiende desde el residuo 90 hasta el 137 y una región de unión flexible entre ambos (residuos del 66 al 89) que permite la actuación de forma independiente de ambos dominios (8-11), y es capaz de interaccionar con otras proteínas. En el dominio N-Terminal recae la capacidad de dimerización/oligomerización e interacciones heteroméricas, mientras que en el dominio C-Terminal recae la capacidad de unión a ácidos nucleicos.
La estructura tridimensional de ambos dominios por separado ha sido resuelta, pero no la estructura tridimensional de la proteína completa, cuya resolución es probablemente impedida por la flexibilidad de la región conectora entre ambos dominios, Figura 1.
Figura 1. Representación esquemática de los dominios funcionales de la proteína H-NS.
El dominio de oligomerización de la proteína H-NS le permite interactuar no sólo con otra copia de sí misma sino también con otras proteínas que muestren este mismo dominio (8). H-NS64 es una forma truncada de H-NS que comprende los 64 primeros residuos. La proteína Hha (High hemolytic activity) fue identificada como modulador de la expresión de la alfa-hemolisina en E. coli (13). Es una proteína de tamaño pequeño (aproximadamente 8,6 kDa y 72 aminoácidos) (6) y con propiedades hidrofílicas. Su estructura ha sido resuelta por RMN (14), mostrando que está formada por 4 hélices alfa que conforman motivos hélice-giro-hélice, Figura 2a.
En cuanto a los niveles de expresión, Hha es una proteína poco abundante en la célula bacteriana y su nivel de expresión es más elevado en fase exponencial que en fase estacionaria. A pesar de que Hha no tiene un dominio de unión a ADN, sí interactúa con H-NS y modula los efectos (1). Estudios sobre la proteína Hha han determinado que su secuencia aminoacídica corresponde a un único dominio funcional de unión a proteínas, mostrando secuencias conservadas con el dominio de oligomerización de H-NS (15).
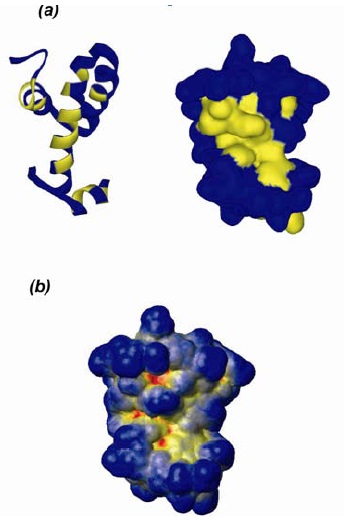
Figura 2. (a). Los residuos de Hha más afectados por la unión H-NS64 son puestos en amarillo. (b) Accesibilidad al disolvente de Hha. Los residuos son coloreados de acuerdo al porcentaje de disolvente expuesto en la superficie: rojo (<5%), amarillo (5 a 20%) y azul (>20%).
Además, la sustitución de este dominio N-Terminal de la proteína H-NS por la secuencia completa de la proteína Hha genera una proteína quimérica funcional, capaz de complementar algunos de los fenotipos de un mutante hns (16).
La proteína HhaC18I es un mutante de Hha, que tiene algunas características que la diferencian y la hacen incluso mejor que otros mutantes y Hha wild type. La mutación C18I no cambia la proteína a nivel estructural, no cambia las propiedades de unión a H-NS, impide que la proteína se reprima y no disminuye el crecimiento celular (17). Anteriormente en nuestro grupo de investigación se comprobó que Hha interacciona únicamente con el dominio N-Terminal de H-NS. Como se puede observar en la Figura 2b se demostró que Hha muestra una plasticidad conformacional ya que los residuos hidrofóbicos son afectados por la interacción con H-NS.
A diferencia de la mayor parte de proteínas asociadas al nucleoide, la proteína H-NS presenta un punto isoeléctrico neutro, pero contiene agrupaciones de aminoácidos con carga que permiten su unión al ADN (19). Las mediciones basadas en la fluorescencia intrínseca del triptófano de Hha, que se obtienen con la anisotropía de fluorescencia, permiten evaluar la afinidad de Hha hacia H-NS. En la literatura ha sido reportado que en E. coli Hha interacciona con H-NS y este complejo interacciona con ADN (20).
En el pasado el grupo aprovechó la fluorescencia intrínseca del triptófano de Hha, que entre los aminoácidos fluorescentes es el mejor para caracterizar la interacción del complejo Hha/H-NS, con el propósito de establecer la relación estructura-función y clarificar el mecanismo de unión y el funcionamiento del complejo. Pero debido a su bajo rendimiento fluorescente, se decidió entonces continuar con el estudio de interacción del complejo Hha/H-NS, mediante la incorporación química de la sonda fluorescente FlAsH en la proteína Hha, con el objetivo de conseguir una mejor sensibilidad. Si esto se logra, en el futuro se podrían hacer estudios de fenómenos de interacción in vitro y también aprovechar la permeabilidad FlAsH respecto a las células bacterianas para estudios in vivo.
La sonda fluorescente FlAsH (fluorescein arsenical helix binder) es un derivado del arsénico, utilizado en proteínas de forma selectiva para observar algunas estructuras, Figura 3. Proporciona una alta sensibilidad y gran versatilidad, con una mínima perturbación en la proteína (21). Tiene interacción en dos puntos con la proteína y por lo tanto poca movilidad, permite la permeabilidad de las membranas, y trabaja con una concentración menor a la que se trabaja con el triptófano y por lo tanto evita la formación de oligomeros.
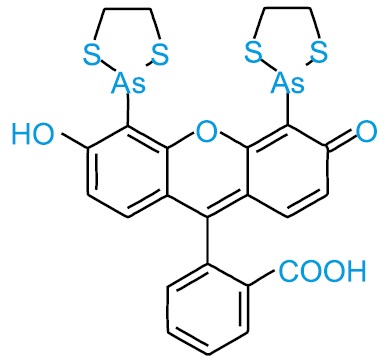
Figura 3. Fluorescein arsenical helix binder (FlAsH).
Según la literatura se sabe que FlAsH sólo fluorese si se tiene en la proteína (en nuestro caso Hha) un vinculo de alta afinidad, un pequeño receptor compuesto por seis aminoácidos (CCPGCC) (22). En el momento en que el FlAsH se une con la proteína su fluorescencia aumenta, por lo tanto es adecuado para el marcado, identificación y estudio estructural de proteínas. FlAsH se incorporará a HhaCCPGCC teniendo en cuenta características de la proteína como absorbancia, peso, concentración, entre otras, y con las precauciones necesarias para evitar la formación de puentes disulfuro y la tendencia a formar especies de alto peso molecular (21).
Para el crecimiento se usaron medios de cultivo líquidos Luria Bertoni (LB), Super Optimal Broth with Catabolite repression (SOC) y sólidos Luria Bertoni/Agar
(LB-Agar).
Antibióticos
Los medios de cultivo fueron suplementados con Ampicilina.
Esterilización
Los medios de cultivo, el material de vidrio y algunas soluciones fueron esterilizadas en autoclave.
Las soluciones que por su composición no permitían su esterilización por este método, fueron esterilizadas mediante filtración a través de filtros estériles de 0,22μm de diámetro de poro (Millipore).
Incorporación de la secuencia CCPGCC
La proteína Hha fue modificada mediante tecnología recombinante con la incorporación de la secuencia “CCPGCC”, los aminoácidos Prolina (P) y Glicina (G), son los preferidos en la actualidad ya que con estos dos aminoácidos se ha demostrado, mediante estudios cinéticos, que esta secuencia tiene mejor estabilidad y están optimizados para una mayor afinidad con el FlAsH (23).
Se hicieron dos construcciones para Hha: HhaCCPGCC1 y HhaCCPGCC2 iguales al comienzo y al final pero con la diferencia que HhaCCPGCC2 tiene una deleción de tres aminoácidos a partir de la metionina inicial (MSE), esta deleción se hizo para obtener una mayor proximidad entre Hha y la sonda fluorescente FlAsH con el objetivo de restringir la dinámica del complejo.
Cualquier incremento en el tiempo de correlación, debido a un proceso de asociación, se observaría en el tiempo de correlación de FlAsH.
Las construcciones se hicieron por medio de procedimientos de PCR con posterior purificación utilizando el kit QIAquick PCR purification® (QIAGEN) siguiendo el protocolo recomendado. Luego se realizaron los protocolos de digestión y de ligación usando el rapid DNA ligation kit® (ROCHE) siguiendo el protocolo recomendado.
Mutagénesis
En la obtención del mutante HhaC18I se utilizó el kit QuickChange® XL Site-Directed (Stratagene) siguiendo el protocolo recomendado. La metodología de mutagénesis consistió en amplificar mediante PCR el vector de expresión que contiene el gen de Hha wild type, utilizando como cebadores dos oligonucleótidos con la mutación deseada, cada uno complementario a cada una de las hebras de DNA que codifica la proteína a mutar. El producto de amplificación se trató con la endonucleasa Dpn I para eliminar el DNA molde utilizado en la PCR. El plásmido mutado y con extremos cohesivos resultante es posteriormente transformado en células competentes XL10-Gold que reparan los extremos del plásmido circularizándolo.
Transformación
El protocolo de transformación utilizado consta de tres pasos: choque térmico, recuperación y sembrado. En el choque térmico, se incubaron células BL21-DE3 con el plásmido que se quería introducir durante 30 minutos a 0ºC, 40 segundos a 42ºC y 2 minutos más a 0º C. Luego se añadió medio SOC y se incubaron a 37ºC durante una hora. Posteriormente, se sembraron las células en una placa LB-Agar con ampicilina y se dejaron a 37ºC durante toda la noche.
Amplificación de fragmentos de ADN mediante la reacción en cadena de la polimerasa (PCR)
Esta técnica fue utilizada para replicar hebras de ADN, para lo cual se emplearon ciclos de altas y bajas temperaturas alternadas para separar las hebras recién formadas entre sí tras cada fase de replicación y luego dejar que se volvieran a unir a polimerasas para duplicarlas. Para realizar la técnica se utilizaron: desoxinucleótidos trifosfato (dNTPs), dos primers, cada uno complementario a una de las dos hebras del ADN, un ión divalente, que en nuestro caso es el cloruro de magnesio (MgCl2), un buffer que mantuvo el pH adecuado, ADN polimerasa y un ADN molde que contiene la región de ADN que se va a amplificar. Las variables de la reacción de amplificación (temperatura de hibridación, tiempos y número de ciclos) se determinaron en función de las características del fragmento de ADN a amplificar y de los primers utilizados. Los productos resultantes fueron analizados mediante electroforesis en geles de agarosa.
Geles agarosa
Los ácidos nucleicos presentes en las muestras se analizaron mediante electroforesis en geles de agarosa que permiten la separación en función de su peso molecular y forma.
Expresión y purificación de las proteínas
Es necesario obtener las proteínas en gran cantidad y con alto nivel de pureza. El proceso se realizó por “expresión de la proteína recombinante” y posterior purificación mediante columna de afinidad. La expresión de proteínas recombinantes se basa en que las bacterias, a parte de su propio cromosoma, pueden albergar en su citoplasma moléculas circulares de ADN que se replican de forma independiente de la del cromosoma bacteriano, llamados plásmidos, que son de tamaño pequeño, fáciles de aislar y permiten introducir insertos de ADN sin afectar la viabilidad de la célula (24).
Las bacterias transformadas con el plásmido correspondiente, el día anterior se hicieron crecer en medio LB a 37º C hasta una densidad óptica a 600nm de 0.6-0.7. En este punto se indujo la expresión con IPTG y se disminuyó la temperatura a 15ºC y se dejaron durante una noche.
Purificación
La purificación consta de los siguientes pasos: se centrifugó el medio de cultivo a 3250g, 30 minutos a 4º C, separando las bacterias del medio de cultivo.
Luego se resuspendieron las células en tampón 20 mM Tris, 800mM NaCl, 5 mM Imidazol, 2mM TCEP. pH=8, para posteriormente congelarlas a -20ºC. Una vez descongeladas las células, se lisaron mediante ultrasonicación en un baño de hielo, alternando seis ciclos de 10 segundos con periodos de 10 segundos de reposo.
Para eliminar la fracción soluble del lisado, se centrifugó a 40000 g por 20 minutos a 4º C. El sobrenadante se incubó con resina Ni-NTA agarosa (QUIAGEN) a temperatura ambiente, la resina se lavó con el tampón de lisis aproximadamente 20 mL, se eluyó a proteína con imidazol en mayor concentración. En el paso final de la purificación la proteína se inyectó en una columna de exclusión molecular Superdex 75 (Amersham) y se eluyó con el tampón correspondiente, Figura 4.
Identificación de proteínas por MALDI-TOF
Con esta técnica se obtiene información sobre la masa de las especies que constituyen la solución proteíca. Si en el espectro se observa un solo pico se dice que es una solución monodispersa, lo que significa que es consistente con la muestra; si se observan varios picos hay identidades con diferentes pesos moleculares en la solución. Este espectro total es el obtenido por un método que utiliza una fuente de ionización y la deserción de muestras asistida por un láser.
Geles de poliacrilamida
Las proteínas presentes en una muestra se analizaron mediante electroforesis en geles desnaturalizantes de poliacrilamida, que permiten la separación en función de su peso molecular.
Protocolo incorporación FlAsH
Para la incorporación del FlAsH se limpió un tubo concentrador centrifugándolo a 3000g por 10 minutos con agua Mili Q y posteriormente con buffer 20 mM NaP, 200mM NaCl, 1mM TCEP, 0.01% NaN3, 0.2mM EDTA, pH 7, luego se añadió la proteína al tubo concentrador y se centrifugó 3000g hasta llegar a una cantidad de muestra de aproximadamente 1 mL, y se dejó a -20ºC. Se encontró la concentración de la proteína para saber que cantidad de FlAsH añadir.
Luego de añadir FlAsH, la muestra se protegió de la luz, y se dejó toda la noche en la noria a temperatura ambiente, al terminar se guardó a -20ºC. Se limpió una columna de desalinización alternando: agua, NaOH, agua, etanol, agua y buffer 20 mM NaP, 150mM NaCl, 1mM TCEP, 0.01% NaN3, 0.2mM EDTA, pH 7.
Luego se pasó a la columna 0.5 mL de buffer + 1.5 mL de muestra y luego se eluyó con el mismo buffer.
Se prepararon cuatro muestras:
1. 0.1 μM FlAsH-EDT2+1μM proteína (HhaCCPGCC1 o HhaCCPGCC2)+ 1μM BAL + 1mM b-mercaptoetanol.
2. 0.05 μM FlAsH-EDT2+1μM proteína (HhaCCPGCC1 o HhaCCPGCC2)+ 1μM BAL + 1mM b-mercaptoetanol.
3. 0.24 μM FlAsH-EDT2+0.1μM proteína (HhaCCPGCC1 o HhaCCPGCC2)+ 1μM BAL + 1mM b-mercaptoetanol.
4. 0.020 μM FlAsH-EDT2+1μM proteína (HhaCCPGCC1 o HhaCCPGCC2)+ 1μM BAL + 1mM b-mercaptoetanol.
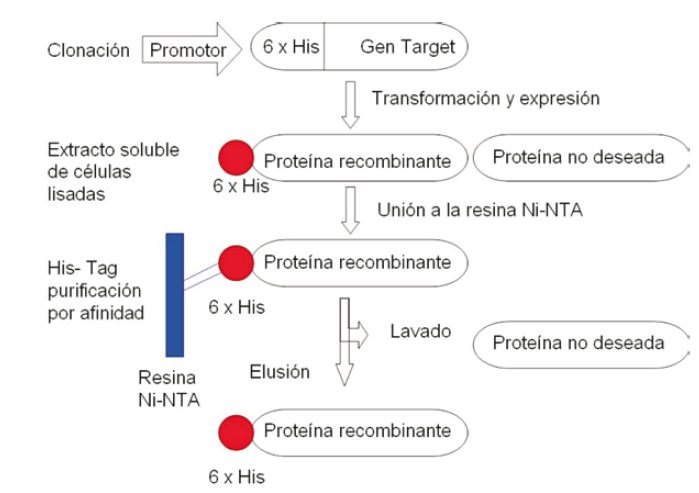 Figura 4. Representación esquemática de la propuesta de purificación.
Figura 4. Representación esquemática de la propuesta de purificación.
Estudios de fluorescencia
Los experimentos de fluorescencia fueron realizados con un espectrofluorímetro, equipado con un autopolarizador con geometría “T”. La excitación para HhaCCPCGG1/FlAsH se realizó a 508 nm y la emisión a 528 nm. Las medidas fueron obtenidas a una temperatura constante de 25ºC. Las medidas de fluorescencia fueron llevadas a cabo en presencia del buffer 20 mM NaP, 200mM NaCl, 1mM TCEP, 0.01% NaN3, 0.2mM EDTA. Las muestras de Hha usadas para las medidas fueron tomadas directamente del monómero obtenido en la purificación por extracción molecular.
Los experimentos de anisotropía de fluorescencia se llevaron a cabo con una emisión y excitación de 5nm para todas las medidas. La anisotropía A fue calculada de acuerdo a la ecuación:
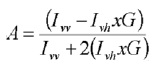
Donde
![]()
Factor de corrección.
Ivv es la medida de la fluorescencia emitida in el plano paralelo al plano de excitación.
Ivh es la medida de la fluorescencia emitida en el plano perpendicular al plano de excitación.
Sobre la muestra 3 se realizaron sucesivas titraciones de H-NS64 purificado con tres concentraciones diferentes a 25ºC.
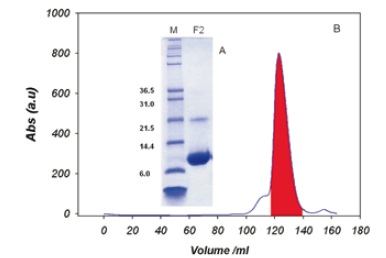
Figura 5. (a) Análisis de la purificación de HhaCCPGCC2 (F2) mediante gel de poliacrilamida. (b) Purificación de la proteína HhaCCPGCC2 por columna de afinidad.
Esto se comprobó mediante Maldi tof, y se puede concluir que se obtuvo HhaCCPGCC con alto nivel de pureza. Para comprobar el estado de oligomerización de Hha se hizo una columna de afinidad. En el perfil de cromatografía se observó un pico mayoritario que corresponde a la forma monomérica de Hha.
Cuando se une el FlAsH a HhaCCPGCC1 empieza la emisión, y se puede observar el incremento de la fluorescencia, así se puede monitorizar la incorporación.
Ya que la incorporación de FlAsH a la secuencia CCPGCC es seguida por un incremento de la fluorescencia, este incremento se siguió en función del tiempo, con una cinética de incorporación a determinadas condiciones experimentales, asegurando que la incorporación llegara a un equilibrio. Cuando las curvas 1, 2 y 4 llegan al plateau se dice que hay una incorporación total del 100%, ya que se tienen las mejores condiciones, Figura 6a.
Si la conversión fuera a 100% en la curva 4, el plateau de esta curva tendría que coincidir con el plateau de la curva 1, ya que el reactivo limitante es igual. Para hallar el porcentaje de incorporación de FlAsH en HhaCCPGCC1 en la curva 3 se hizo una representación, Figura 6b, de las curvas 1, 2 y 4 en función de los valores máximos alcanzados en el plateau. Con la ecuación de la recta se encontró que hay una incorporación del 45% de FlAsH/HhaCCPGCCC1, lo que significa que hay un 55% de HhaCCPGCC1 libre que puede interaccionar con H-NS64 y si se dá esta interacción no se puede observar ningún comportamiento de esta fracción ya que no habrá fluorescencia. La contribución del complejo HhaCCPGCC1/FlAsH (45%) va a ser aproximadamente la misma contribución de HhaCCPGCC1, lo cual no es un buen resultado.
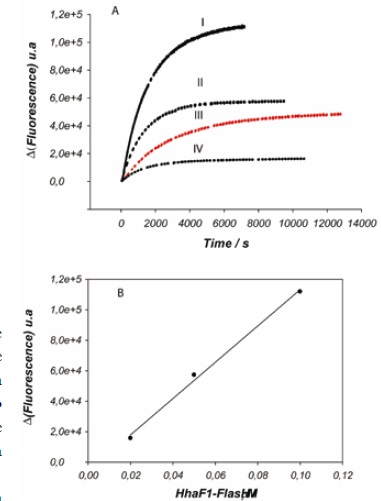
Figura 6: (a) Representación de las incorporaciones de FlAsH respecto a HhaCCPGCC1. La curva 3 muestra el cambio en la fluorescencia causado por la adición de H-NS. (b) Representación de las curvas 1,2 y 4 en función de sus valores máximos alcanzados en el plateau.
Se tienen varias hipótesis para explicar el por qué la incorporación del FlAsH no se dió al 100%. No se trabajó con un rango de concentraciones con exceso de FlAsH; si se tiene mayor cantidad de FlAsH (por ejemplo 1μM) que de proteína (por ejemplo 0.1μM) se garantiza una interacción del 100%, pero esto no se hizo por razones económicas. Además, no tendría sentido hacerlo porque al aumentar la concentración de FlAsH, este quedaría con concentraciones similares a las que se tenían cuando se trabajaba con el triptófano, y la idea es disminuir estas concentraciones y gastar menos cantidad de proteína.
El mercaptoetanol compite con los arsénicos del FlAsH por las cisteinas de la secuencia CCPGCC, y al tener mayor cantidad de mercaptoetanol que de proteína se puede llegar a afectar la incorporación proteína/FlAsH. BAL y la proteína compiten por el arsénico del FlAsH y a pesar de que la proteína une mejor que el BAL como se trabajó con un exceso (0.1 μM HhaCCPGCC1 y 1μM BAL) esto podría afectar la incorporación proteína/Flash. Estos detalles se deben optimizar con más experimentos. Tal vez la proteína y la secuencia CCPGCC no forman un complejo 100% funcional y esto impide una buena incorporación.
La Figura 7 muestra el cambio en la anisotropía de fluorescencia de Hha causado por la adición de H-NS64.
Se intentó ver un cambio entre el estado libre de la proteína y el estado acomplejado, pero no se observó un cambio notable hasta aproximadamente el rango de 10 μM, donde la anisotropía de fluorescencia de Hha empieza a incrementar con las adiciones de H-NS64. El hecho de no observar cambios desde el comienzo se pudo dar por diferentes motivos. Las concentraciones bajas posiblemente no favorecen la reacción. Las condiciones de sal con las cuales se trabajó (150mM) son altas, por lo tanto no hay buena afinidad. Posiblemente el linker es muy grande o estaba rígido, afectando notablemente la interacción.
Los datos obtenidos permiten decir que se logró construir la proteína HhaCCPGCC capaz de unir la sonda fluorescente FlAsH y además de eso se evidenció la estabilidad, buen rendimiento y pureza a pesar de tener una secuencia artificial.
Esta construcción se logró sin problemas. Aunque los estudios de interacción y caracterización estructural entre las proteínas HhaCCPGCC y H-NS64 mediante anisotropía de fluorescencia no se pudieron llevar a cabo, este informe muestra algunas hipótesis acerca del por qué algunos detalles no se resolvieron y partiendo de ellos y de los resultados obtenidos se pueden desarrollar trabajos futuros.
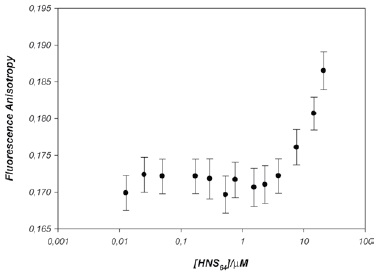
Figura 7. Representación de las titraciones de H-NS64 sobre HhaCCPGCC1/FlAsH.
1. Madrid C, Balsalobre C, García J, Juárez A. The novel Hha/YmoA family of nucleoid-associated proteins: use of structural mimicry to modulate the activity of the H-NS famil of proteins. Mol Microbiol. 2007;63:7 14.
2. Varshavsky, A., Nedospasov, S. A., Bakayev, V. V., Bakayeva, T. G. and Georgiev, G. P. Histone-like proteins in the E. coli chromosome. Nucl. Acids Res. 1977:4: 2725-2745.
3. Lammi M, Paci M, Pon CL, Losso MA, Miano A, Pawlik RT, et al. Proteins from the prokaryotic nucleoid: biochemical and 1H NMR studies on three bacterial histone-like proteins. Adv Exp Med Biol. 1984;179:467-477.
4. Spassky A, Rimsky S, Garreau H, Buc H. H1a, an E. coli ADN-binding protein which accumulates in stationary phase, strongly compacts AND in Vitro. Nucleic Acids Res. 1984;12.5321-5340.
5. Schröder O, Wagner R. The bacterial regulatory protein H-NS- -a versatile modulator of nucleic acid structures. Biol Chem. 2002;383:945-960.
6. Pons I. La familia de proteínas Hha-YmoA: estudios estructurales y papel regulador en Yersinia Enterocolitica. Tesis Doctoral. Universitat de Barcelona, 2006.
7. Azam TA, Ishihama A. Twelve species of the nucleoid-associated protein from Escherichia coli. Sequence recognition specificity and ADN binding affinity. J Biol Chem. 1999;274:33105-33113.
8. Dorman CJ, Hinton JC, Free A. Domain organization and oligomerization among H-NS-like nucleoid associated proteins in bacteria. Trends Microbiol. 1999;7:124-128.
9. Badaut C, Williams R, Arluison V, Bouffartigues E, Robert B, Buc H, Rimsky S. The degree of oligomerization of the H-NS nucleoid structuring protein is related to specific binding to ADN. J Biol Chem. 2002;277:41657-41666.
10. Esposito D, Petrovic A, Harris R, Ono S, Eccleston JF, Mbabaali A, et al. H-NS oligomerization domain structure reveals the mechanism for high order self-association of the intact protein. J Mol Biol. 2002;324:841- 850.
11. Bloch V, Yang Y, Margeat E, Chavanieu A, Augé MT, Robert B, et al. The H-NS dimerization domain defines a new fold contributing to AD recognition. Nat Struct Biol. 2003;10:212-218.
12. Dorman CJ. H-NS: A universal regulador for a dynamic genome. Nat Rev Microbiol 2004;12:179-184.
13. Godessart N, Muñoa FJ, Regue M, Juárez A. Chromosomal mutations that increase the production of a plasmid-encoded haemolysin in Escherichia coli. J Gen Microbiol. 1988;134:2779-2787.
14. Yee A, Chang X, Pineda-Lucena A, Wu B, Semesi A, Le B, et al. An NMR approach to structural proteomics. Proc Natl Acad Sci U S A. 2002;99:1825-1830.
15. Nieto JM, Madrid C, Miquelay E, Parra JL, Rodríguez S, Juárez A. Evidence for direct protein-protein interaction between members of the enterobacterial Hha/YmoA and H-NS families of proteins. J Bacteriol. 2002;184:629-635.
16. Rodríguez S. Interacción entre proteínas de las familias H-NS y Hha / YmoA: regulación de la expresión génica en Enterobacteriaceae. Tesis Doctoral. Facultad de Biología. Universitat de Barcelona, 2005.
17. Cordeiro T. Dynamic and structural aspects of the interaction between Hha and H-NS. Diploma of Avanced Studies in Organic Chemistry. Universitat de Barcelona- IRB- Barcelona Science Park, 2008.
18. García J, Cordeiro TN, Nieto JM, Pons I, Juárez A, Pons M. Interaction between the bacterial nucleoid associated proteins Hha and H-NS involves a conformational change of Hha. Biochem J. 2005;388:755-762.
19. Rimsky S. Structure of the histone-like protein H-NS and its role in regulation and genome superstructure. Curr Opin Microbiol. 2004;7:109- 114
20. Cordeiro T. Insights into the Interaction between Nucleoid-associated Proteins, Hha and H-NS. Formal Training Report. Platform Biomolecular RMN , Barcelona Science Park, Universitat de Barcelona. 2005.
21. Zhang J, Campbell RE, Ting AY, Tsien RY. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906-918.
22. Stroffekova K, Proenza C, Beam KG. The protein-labeling reagent FLASH-EDT2 binds not only to CCXXCC motifs but also nonspecifically to endogenous cysteine-rich proteins. Pflugers Arch. 2001;442:859-866.
23. Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J Am Chem Soc. 2002;124:6063-6076.
24. Fernández De Alba, C. Estudio de la interacción entre proteínas asociadas al nucleoide mediante anisotropía de fluorescencia. Máster en Química orgánica experimental. Universitat de Barcelona, Institut de Recerca Biomédica de Barcelona, 2007