Oxidative stress-induced mitochondrial failure and vasoactive substances as key initiators of pathology favor the reclassification of Alzheimer Disease as a vasocognopathy Ludis Morales Alvarez1, Gerardo Jesus Pacheco2,3, Hector Palacios2,3, Brianna Walrafen2,3, Mark Eric Obrenovich4, Eldar Qasimov5, Joseph Charles LaManna6, and Gjumrakch Aliev1,2,3
1Department of Nutrition and Biochemistry, Javeriana University, Bogota, Colombia
2Department of Biology, and 3 Electron Microscopy Research Center, College of Sciences, University of Texas at San Antonio, San Antonio, Texas 78249, USA;4Department of Pathology, Case Western Reserve University, Cleveland, Ohio 44106, USA, 5 Department of Cytology, Histology & Embryology, Azerbaijan Medical University, Baku Azerbaijan,6Department of Anatomy, Case Western Reserve University, Cleveland, Ohio 44106, USA.
Correspondencia: gjumrakch.aliev@utsa.edu, aliev03@gmail.com
Recibido: 17-09-08 / Aceptado: 04-11-08
Alzheimer disease, oxidative stress occurs within various cellular compartments and within certain cell types more than others, namely the vascular endothelium, which is associated with atherosclerotic damage, as well as in pyramidal neurons and glia. Interestingly, these vulnerable cells show mtDNA deletions and oxidative stress markers only in the regions that are closely associated with damaged vessels. This evidence strongly suggests that chronic hypoperfusion induces the accumulation of the oxidative stress products. Furthermore, brain vascular wall lesions linearly correlate with the degree of neuronal and glial cell damage. We, therefore, conclude that chronic hypoperfusion is a key initiator of oxidative stress in various brain parenchymal cells, and the mitochondria appear to be primary targets for brain damage in Alzheimer disease. In this manuscript, we outline a role for the continuous accumulation of oxidative stress products, such as an abundance of nitric oxide products (via the overexpression of inducible and/or neuronal NO synthase (iNOS and nNOS respectively) and peroxynitrite accumulation, as secondary but accelerating factors compromising the blood brain barrier (BBB). If this turns out to be the case, pharmacological interventions that target chronic hypoperfusion might ameliorate the key features of dementing neurodegeneration.
Key words: brain hypoperfusion, mitochondria, neurodegeneration, nitric oxide, oxidation, oxidative stress, vasoactive substances.
La enfermedad de Alzheimer y el accidente cerebrovascular son dos causas que conducen a la demencia relativa a la edad. El aumento de la evidencia apoya la idea de que la hipoperfusión crónica es sobre todo responsable de la patogénesis que es la base de ambos procesos de la enfermedad. La hipoperfusión se asocia al desequilibrio oxidativo, en gran parte debido a las especies reactivas del oxígeno, que se asocian a otros desórdenes degenerativos relativos a la edad. Evidencia reciente indica que un estímulo crónico de lesión induce la hipoperfusión vista en la microcirculación de las regiones vulnerables del cerebro. Esto conduce a la falta de energía, manifestada por daño a la ultraestructura mitocondrial. Los trastornos mitocondriales conducen a la formación de una gran cantidad de mitocondrias “hipoxicas” electrón-densas y causan la sobreproducción de deleciones en el ADN mitocondrial (mtADN), debidas muy probablemente a la ruptura de la doble hélice. Además, estas anormalidades mitocondriales coexisten con la actividad redox incrementada, peroxidación lipídica y la oxidación del ARN, que son características bien establecidas de la patología de Alzheimer, antes de la aparición de los depósitos del b-amiloide.
En la enfermedad de Alzheimer, el estrés oxidativo ocurre dentro de los varios compartimientos celulares y dentro de ciertos tipos de células más que en otros, a saber el endotelio vascular, que se asocia a daño aterosclerótico, así como en las neuronas piramidales y glia. Es interesante que estas células vulnerables demuestran deleciones del mtADN y marcadores de estrés oxidativo solamente en las regiones que están cercanamente relacionadas a los vasos dañados. Esta evidencia sugiere fuertemente que la hipoperfusión crónica induce la acumulación de los productos del estrés oxidativo. Además, las lesiones de la pared vascular del cerebro se correlacionan linealmente con el grado de daño celular neuronal y glial. Por lo tanto, concluimos que la hipoperfusión crónica es un iniciador clave del estrés oxidativo en varias células parénquimales del cerebro, y las mitocondrias parecen ser blancos primarios para el daño de cerebro en la enfermedad de Alzheimer. En este trabajo, delineamos el papel de la acumulación continua de los productos del estrés oxidativo, tales como una abundancia de productos de óxido nítrico (vía la sobreexpresión de NO sintasa inducible y/o neuronal (iNOS y nNOS respectivamente) y de acumulación del peroxinitrito, como factores secundarios pero acelerantes que comprometen la barrera hematoencefálica. Si éste resulta ser el caso, las intervenciones farmacológicas que apuntan a la hipoperfusion crónica pudieran mejorar las características dominantes de la demencia neurodegenerativa.
Palabras clave: estrés oxidativo, hipoperfusión cerebral, mitocondria, neurodegeneración, oxidación, óxido nítrico, sustancias vasoactivas.
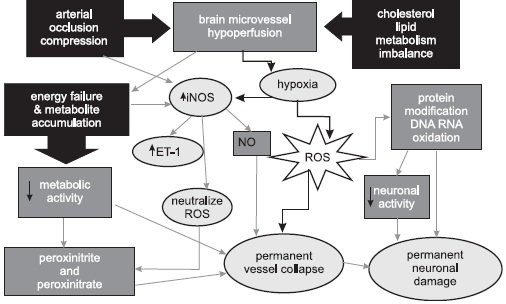
Diagram 1. Possible pathogenetic mechanisms of the Effect of Vascular hypoperfusion during aging and the development of stroke and AD. Common relationships between AD and the cardiovascular and cerebrovascular diseases (reprinted with permission of Neurotoxicity Research, 2006 (10(1):43-56).
The three main flaws of the hypothesis appear to be: 1. Ab deposition has not been found to be toxic or to cause the damage and death of cerebrally located nerve cells in humans or animals; 2. the brains of many aged, but cognitively normal individuals show abundant Ab-containing senile plaques but no clinical signs of Alzheimer’s disease; and 3. since there is general agreement that Ab-containing senile plaques are the products of degenerating neurons, they can not be the cause, since it is axiomatic that a product is the result, not the cause, of some activity.
Considerable evidence, in contrast, indicates that late onset AD is a vascular disorder whose underlying cause is impaired blood flow to the brain during advanced aging. This evidence can be summarized as follows: 1. numerous epidemiological studies link AD risk factors such as stroke, heart disease, hypertension, and atherosclerosis to reduced cerebral blood flow; 2. evidence that AD and vascular dementia (VaD), an acknowledged vascular disorder, share practically all the same risk factors and may benefit from the same treatments; 3. drug therapies reported to improve AD symptoms (including prescriptive drugs now available for AD) all increase blood flow to the brain; 4. people who are likely to develop AD but do not yet show dementia symptoms can be identified by using brain blood flow measurements and brain PET scans; 5. the clinical symptoms are very similar in most AD and VaD patients; 6. parallel abnormalities such as Ab-laden plaques found in AD and VaD patients occur in both brain vessels and brain tissue; 7. low levels of brain blood flow in aged humans and animal models can lead to abnormal cell metabolism, tissue damage, and memory problems, independent of Ab; 8. mild cognitive impairment (a term used to describe a preliminary stage leading to AD) can convert equally to AD or VaD; and 9. small vessel damage is also present in the majority of AD brains after death.
For these reasons, it is suggested that AD be reclassified as a vascular disorder and described as a “vasocognopathy”. The term aptly describes the origin of the disease (vaso: vessel blood flow), its primary effect on a system (-cogno: relating to mental function), and its clinical course (-pathy: disorder). Reclassification of AD from a neurodegenerative to a vascular disorder would speed the development of truly beneficial treatments or a cure, improve patient management, provide earlier diagnosis, and reduce the number of AD cases in the future by aggressively treating the risk factors that can promote this dementia. The literature reveals no compelling evidence that Ab deposition causes AD or that it results in significant damage to brain cells. By contrast, the findings that support AD as primarily a vascular disorder are substantially more compelling. Determining the underlying mechanisms behind vascular defects in experimental animals will provide crucial information in the development of new, more effective therapies for the treatment of atherosclerosis, including cerebrovascular athero- and arteriosclerosis. Because the cerebrovascular pathology found in mild cognitive impairment (MCI) and AD leads to mental deterioration and progressive neurodegeneration, the mechanism of its formation deserves special attention. Therefore, pharmacological intervention aimed at correcting chronic brain hypoperfusion will also be useful for treating and preventing dementing neurodegeneration, Diagram 1.
Whereas the pathogenesis of atherosclerosis has been intensively studied and described, the underlying events that initiate cardiovascular disease are not yet fully understood. A substantial number of studies suggest that altered levels of oxidative and nitrosoxidative stress within the cardiovascular environment are essential in the development of cardiovascular disease; however, the impact of such changes on organelles and other subcellular components and their functions that are relevant to cardiovascular disease inception are less understood. In this regard, studies are beginning to show that mitochondria not only appear susceptible to damage mediated by increased oxidative and nitrosoxidative stress, but also play significant roles in the regulation of cardiovascular cell function. In addition, accumulating evidence suggests that a common theme between cardiovascular disease development and cardiovascular disease risk factors is increased mitochondrial damage and dysfunction (1-5).
The vascular endothelium, which regulates the passage of macromolecules and extravasation of immune cells from blood into tissues, is a major target of oxidant stress and plays a critical role in the pathophysiology of several vascular diseases (2,3,5). Reactive oxygen species (ROS) are generated at sites of injury and/or inflammation. Interestingly, the vascular endothelium, neurons, and glia are all able to synthesize, store, and release ROS as well as vasoactive substances in response to certain stimuli, especially chronic hypoxia/hypoperfusion. The contribution of ROS to the pathophysiology of stroke, cerebrovascular disease, and AD is extremely important. Moreover, hypoperfusion, as an underlying cause of oxidative-stress producing vascular lesions, is a promising avenue for determining the etiopathogenesis of AD (6,7).
This notion is based on a positive correlation between AD and cardiovascular diseases. Specifically, accumulated oxidative stress increases vascular endothelial permeability and promotes leukocyte adhesion that is coupled to alterations in endothelial cell signal transduction and redox-regulated transcription factors. It seems highly probable that the cellular and molecular mechanisms by which hypoperfusion-induced ROS accumulation impairs endothelial barrier function and promotes leukocyte adhesion could result in the development of AD. Sustained hypoperfusion and oxidative stress of brain tissues could also stimulate the secondary overexpression of inducible and neuronal-specific nitric oxide synthase (NOS: iNOS and nNOS, respectively) and endothelin-1 (ET-1) in brain cells. Also, increased accumulation of oxidative stress products probably contributes to both the decompensation of the BBB and damage to brain parenchymal cells (2,3-5).
Many common underlying risk factors play key roles in the development of cardiovascular, cerebrovascular, and neurodegenerative diseases (6,8,9). For example, tobacco smoking is accepted as a risk factor for the development of cancer as well as cardiovascular, cerebrovascular, and pulmonary diseases. In addition, cigarette smoking indirectly leads to the formation of free oxygen radicals by inducing chronic hypoxic conditions. In support of this concept, new evidence indicates that continuous formation of free oxygen radicals induces cellular damage and leads to a reduction in cytoprotective mechanisms (9-12). Several recent studies show that cigarette smoking is a cofactor in the initiation of AD, via its effect on the vasculature. Vascular insufficiency/hypoperfusion is a pathogenetic factor in the development of cerebrovascular diseases, such as stroke and especially cerebrovascular atherosclerosis, indicating the latter may also be linked to the pathogenesis of AD. However, the role of tobacco smoking in the pathogenesis of AD is still unclear and controversial.
Cerebral-vascular energy requirements: Hypoperfusion as a key factor for the development of AD
The role of hypoperfusion-induced oxidative stress in vascular abnormalities has been demonstrated in the pathogenesis of AD. Several studies have shown chronic cerebral hypoperfusion in AD and concluded that it is secondary to a reduction in the need for oxygen (7,13-16). However, a greater fraction of oxygen is removed from the vasculature in AD patients as compared to non-AD controls (17). This suggests that low vascular blood flow is a prominent feature of the brain during chronic hypoxia/hypoperfusion and may be a main initiating factor during the development of AD (18,19).
AD is characterized by the impairment of brain energy metabolism (20). Positron emission tomography (PET) has revealed a decline in the cerebral metabolic rate of the parietal and temporal lobes during AD (21,22). These metabolic defects are present before AD symptoms develop in apolipoprotein E (ApoE) e4 homozygous patients (22). De la Torre (18) proposes that advanced aging in conjunction with a comorbid condition, such as a vascular risk factor that further decreases cerebral perfusion, promotes a critically attained threshold of cerebral hypoperfusion (CATCH). With time, CATCH induces brain capillary degeneration and suboptimal delivery of energy substrates to neuronal tissue (18).
Since glucose is the main fuel of brain cells, its impaired delivery, together with a deficient delivery of oxygen, compromises neuronal stability because the supplies for aerobic glycolysis fail to meet brain tissue demand. The outcome of CATCH is a metabolic cascade that involves, among other things, mitochondrial dysfunction, oxidative stress, decreased adenosine triphosphate (ATP) production, increased calcium entry, abnormal protein synthesis, cell ionic pump deficiency, signal transduction defects, and neurotransmission failure. These events contribute to the progressive cognitive decline characteristic of patients with AD, as well as to regional anatomic pathology, consisting of synaptic loss, SPs, neurofibrillary tangles (NFTs), tissue atrophy, and neurodegeneration. CATCH identifies the clinical heterogenic pattern that characterizes AD providing compelling evidence that any of a multitude of different etiopathophysiologic vascular risk factors, in the presence of advanced aging, can lead to AD (2,3-5,18,23,24).
The effect of oxidative stress on brain microvessel function and Ab deposition in AD
Reactive oxygen species and oxidative state are slowly gaining acceptance in having a physiological relevance rather than just being the culprits in pathophysiological processes (3,4). The control of the redox environment of the cell provides for additional regulation in relation to critical cellular signal transduction pathways. Conversely, aberrant regulation of oxidative state manifesting as oxidative stress can predispose a cell to an adverse outcome. The PI3-kinase/Akt pathway is one such pathway that is partially regulated via oxidative state and, in an oxidative stress paradigm such as ischemic reperfusion injury, may be inactivated, which can lead to potentiation and/or exacerbation of cell death. Activation of NFkB has also been associated with oxidative stress. The role of NFkB in neuronal cell death is widely debated, with major studies highlighting both a pro- and an antiapoptotic role for NFkB with the outcome being region, stimulus, dose, and duration specific.
This review hopes to make clear that the oxidative state of a cell plays a key role in the regulation and control of its numerous signal transduction pathways and that elucidating the mechanisms behind oxidative stress-mediated neuronal cell death is important in identifying potential putative targets for the treatment of neuropathologies such as stroke (25). Moreover, recently we have reported (5), the role of G protein-coupled receptor kinase 2 (GRK2) as it relates to individuals affected by AD and how the cardiovasculature plays a role in AD pathogenesis. The possible involvement of GRKs in AD pathogenesis is an interesting notion, which may help bridge the gap in our understanding of the heart-brain connection in relation to neurovisceral damage and vascular complications in AD, since kinases of this family are known to regulate numerous receptor functions both in the brain, myocardium, and elsewhere.
Our findings of overexpression of GRK2 in the context of the early pathogenesis of AD, since increased levels of GRK2 immunoreactivity were found in vulnerable neurons of AD patients as well as in a two-vessel occlusion (2-VO) mammalian model of ischemia. Also, we consider the consequences for this overexpression as a loss of G-protein coupled receptor (GPCR) regulation, as well as suggest a potential role for GPCRs and GRKs in a unifying theory of AD pathogenesis, particularly in the context of cerebrovascular disease. We synthesize this newer information and attempt to put it into context with GRKs as regulators of diverse physiological cellular functions that could be appropriate targets for future pharmacological intervention (5).
Reactive oxygen species (ROS) are considered to be chemically reactive with as well as damaging to biomolecules such as DNA, proteins, and lipids. It is also know that excessive exposure to ROS induces oxidative stress and causes genetic mutations (2). However, the recently described family of Nox and Duox enzymes generates ROS in a variety of tissues as part of normal physiological functions, which include innate immunity, signal transduction, and biochemical reactions, e.g., to produce thyroid hormone. Nature’s “choice” of ROS to carry out these biological functions seems odd indeed, given its predisposition to cause molecular damage. This review describes normal biological roles of Nox enzymes as well as pathological conditions that are associated with ROS production by Nox enzymes.
By far the most common conditions associated with Nox-derived ROS are chronic diseases that tend to appear late in life, including atherosclerosis, hypertension, diabetic nephropathy, lung fibrosis, cancer, Alzheimer’s disease, and others. In almost all cases, with the exception of a few rare inherited conditions (e.g., related to innate immunity, gravity perception, and hypothyroidism), diseases are associated with overproduction of ROS by Nox enzymes; this results in oxidative stress that damages tissues over time. We propose that these pathological roles of Nox enzymes can be understood in terms of antagonistic pleiotropy: activated genes that confer a reproductive advantage early in life, but can have harmful effects later in life. Such genes are retained during evolution despite their harmful effects because the force of natural selection declines with advanced age. This review discusses some of the proposed physiologic roles of Nox enzymes, and emphasizes the role of Nox enzymes in disease and the likely beneficial effects of drugs that target Nox enzymes, particularly in chronic diseases associated with an aging population (26).
Recently, reactive oxygen species (ROS) have been found to stimulate an angiogenic response in ischemic reperfused hearts. Short exposure to hypoxia/reoxygenation, either directly or indirectly, produces ROS that induce oxidative stress that is associated with angiogenesis or neovascularization. ROS can cause tissue injury on one hand but promote tissue repair on the other hand by encouraging angiogenesis. It thus appears that after causing injury to the cells, ROS promptly initiate the tissue repair process by triggering an angiogenic response. Angiogenesis is thought to be regulated by several growth factors (EGF, TGF-a, b-FGF, VEGF). Induction of these angiogenic factors is triggered by various stresses. For instance, tissue hypoxia exerts its pro-angiogenic action through various angiogenic factors, the most notable being vascular endothelial growth factor (VEGF), which has been associated with initiating the process of angiogenesis mainly through the recruitment and proliferation of endothelial cells (27).
ROS are generated at sites of inflammation and injury. At low levels, they can function as signaling intermediates in the regulation of fundamental cell activities such as growth and adaptation responses. At higher concentrations, ROS can cause cell injury and death. The vascular endothelium, which regulates the passage of macromolecules and circulating cells from blood to tissue, is a major target of oxidant stress and therefore plays a critical role in the pathophysiology of vascular diseases (28).
Specifically, oxidative stress increases vascular endothelial permeability and promotes leukocyte adhesions that are coupled to alterations in endothelial signal transduction and redox-regulated transcription factors (28). Based on these recent findings, it is hypothesized that ROS impair endothelial barrier function and indirectly induce alterations in normal vascular endothelial cell function which then results in the development and maturation of cerebrovascular disease and AD.
Compared to other organs or tissues, the brain is more vulnerable to ROS-induced damage due to its high rate of oxygen consumption, high polyunsaturated lipid content, and relative paucity of classic antioxidant enzymes (29). Increased levels of oxidative stress in certain brain regions characterize AD (30-33). Studies have demonstrated a decline in polyunsaturated fatty acids (PUFAs) (34-36), as well as an increase in levels of lipid peroxidation (35), protein oxidation (36,37), DNA oxidation (38-41), and RNA oxidation (41-44) during AD. Additionally, oxidative stress markers such as advanced glycation end products (AGEs) and glycoxidative end products (e.g. N-e-carboxy-methyl-lysine and lipid peroxidation adducts) are present in both neurofibrillary tangles (NFTs) and senile plaques (SPs) in AD (37,45-47) and in post-ischemic tissues (48-52).
Vascular aging is associated with both structural and functional changes that can take place at the level of the endothelium, the vascular smooth muscle cells (vSMCs), and the extracellular matrix of blood vessels. In the endothelium, reduced vasodilatation in response to agonists occurs in large conduit arteries and in resistance arteries as a result of aging (53). Furthermore, enhanced oxidative stress by hypoperfusion contributes significantly to the deleterious effects of aging on the endothelium by means of NO breakdown due to ROS. The relative contribution of the above phenomenon to age-related endothelial dysfunction is highly dependent on the species and the type of vascular bed involved (53-56).
One of the hallmark features of AD are Ab deposits present in cortical and subcortical gray matter and in meningeal and gray matter blood vessels (congophilic angiopathy) (57,58). In vivo experimental evidence indicates that these Ab deposits induce cerebrovascular dysfunction in the rat brain (59) and that the Ab peptide produces endothelial dysfunction in cerebral microvessels via ROS. Accumulating evidence also supports the idea that the Ab peptide is responsible for the cerebrovascular effects of amyloid b precursor protein (AbPP) overexpression (60,61). A study by Iadecola and coworkers demonstrated that transgenic mice overexpressing AbPP have a profound and selective impairment in endothelium-dependent regulation of the neocortical microcirculation. This indicates that peptides derived from AbPP processing may contribute to alterations in cerebral blood flow (CBF) and thus neuronal dysfunction during AD (60).
Although amyloid b 1-40 (Ab1-40) did not influence the increasing CBF produced by endothelium-independent vasodilators and hypercapnia, it did contribute to the attenuation of the resting CBF as well as the increasing CBF produced by endothelium-dependent vasodilators. In contrast, Ab1-42 had the exact opposite effect; although it did not lessen the resting CBF or the increasing CBF produced by endothelium-dependent vasodilators, it could influence the increasing CBF produced by endothelium-independent vasodilators and hypercapnia.
The superoxide scavengers SOD and MnTBAP (superoxide dismutase and Manganese (III) tetrakis (4-benzoic acid) porphyrin) reversed the cerebrovascular effects of Ab1-40. This data strongly suggests that Ab1-40, but not Ab1-42, produces the cerebrovascular alterations seen in AbPP transgenic mice, and that Ab1-40 could therefore play a role in the cerebrovascular alterations observed in Alzheimer’s dementia (57,61). Moreover, this study supports the recent evidence demonstrating that brain microvessels isolated from cases of AD have the ability to kill neurons in vitro (62). However, despite all of the research focused on Ab (which, in the later stages of AD progression eventually becomes a source of ROS in vivo (11), the relationship of its effects to hypoperfusion is still not completely understood.
Morphometric and Neuropathological features of cerebrovascular lesions and Ab in AD
Several morphometric features of BBB dysfunction in patients with pathologically confirmed AD have been reported (63). Accumulation of Ab deposits around vessels in AD brain biopsy samples may be an indication of a breach in the BBB during AD progression (63-65). Recent findings strongly support the hypothesis that structural or physiological abnormalities of the BBB itself may represent a seminal pathogenic event during the development of AD (64), thereby leading to vascular amyloid deposition in the brain (34,63,66). The heterogeneous pathology of AD is due to variability in the nature and severity of vascular lesions as well as to its co-existence with cerebrovascular diseases such as cerebrovascular arteriosclerosis (CVA).
For example, significantly higher densities of Ab immunoreactive plaques are present in AD with CVA as compared to AD alone (67). The Ab deposits in SPs and cerebrovascular angiopathy are derived from AbPP expressed in neurons and in a variety of non-neuronal cells (some outside of the central nervous system) (68-72). Perivascular Ab deposition may be a risk factor for reduced regional CBF (rCBF) (73). The age-related losses of mechanisms/cells that are capable of removing Ab deposits involve subtle molecular alterations in components of the basement membrane that allow it to bind Ab and protect it from cellular degradation (74). These alterations, along with the activation of non-neuronal cells such as microglia, further contribute to neuronal damage (75).
Autophagy is mostly a cleanup process and it is particularly important to cells such as neurons. Autophagy also acts as a defense; any foreign organism that evades the extracellular immune system and goes through the cell membrane becomes a potential target for the autophagy system. That is the case with harmful viruses and bacteria. So, it is not strange to think that a malfunctioning autophagy system can be detrimental especially in brain neurons in pathologies like cancer and AD as well during aging itself. In an aging brain neuron, autophagosomes can fail leading to a buildup of damaged proteins and consequent swelling in a neurite, or projection from the cell body of the neuron. The immature autophagosomes collect at the same site. Enzymes that create protein fragments called amyloid b seem to concentrate on the immature autophagosomes (76).
Autophagic vacuoles accumulate abnormally in affected neurons of several major neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease. They have been linked to various aspects of disease pathogenesis including neuronal cell death (77). To determine whether oxidative stress has any influence on the relationship between lysosomes and Ab1–42 (the most toxic form of Ab), the effect of hyperoxia (40% versus 8% ambient oxygen) on the intracellular localization of Ab1–42 was assessed. The study used immunocytochemistry in retinoic acid differentiated SH-SY5Y neuroblastoma cells maintained in serum-free OptiMEM medium. In control cells, Ab1–42 was mainly localized to small non-lysosomal cytoplasmic granules.
Only occasionally Ab1–42 was present in large (over 1 μm) lysosomal-associated membrane protein 2 positive vacuoles, devoid of the early endosomal marker rab5. These large Ab1–42-containing lysosomes were not detectable in the presence of serum (known to suppress autophagy), while their number increased dramatically (up to 24-fold) after exposure of cells to hyperoxia for 5 days. Activation of autophagy by hyperoxia was confirmed by transmission electron microscopy. Furthermore, an inhibitor of autophagic sequestration, 3-methyladenine, prevented the accumulation of Ab1–42-positive lysosomes due to hyperoxia. In parallel experiments, intralysosomal accumulation of Ab1–40 followed oxidative stress as well. The results suggest that Ab can be autophagocytosed and its accumulation within neuronal lysosomes is enhanced by oxidative stress (78). Together these findings suggest that a breakdown in autophagy may contribute to AD.
Several factors that may ameliorate AD have either been associated with improved CBF or have prevented CBF decline (73). The direct relationship between vascular changes in the brain and the pathology of AD is based on ultrastructural studies that reveal widespread penetration of Ab deposits by degenerating microvessels (57,79). However, numerous morphometric studies have demonstrated that endothelial cell (EC) contact with the vast majority of SPs are by chance, and while it is not unusual to show a close proximity in the highly vascular brain (80), there is an actual exclusion of vessels from most SPs. It is also clear that a certain subpopulation of SPs shows a real and intimate relationship with the vasculature (80,81).
It is likely that SPs have more than one origin (82,83), and that vessels are probably integrally involved in the formation of one or more subpopulations. In over 90% of AD cases, Ab can be detected in at least some vessels (84), and the sources of this Ab are likely vascular ECs and SMCs rather than neurons, since ECs and SMCs show an abundant AbPP immunoreactivity (64,80,85,86). Ultrastructural studies on blood vessels containing Ab deposits have shown their intermittent associations with membrane abnormalities of SMCs (82,83). Indeed, in AD cases with a clinical history of cerebral bleeding, the muscle layer is sometimes completely replaced by Abdeposits (80,85,86). This finding suggests that vascular wall cell alterations such as EC damage and muscle cell atrophy may occur in AD, even in the absence of visible Ab depositions, and implies that the vascular system is a primary target for the development of this disease.
Relationships between ApoEgenotype, hypercholesterolemia, Ab and vascular changes in AD
The association of Ab with cerebral vessels is an intriguing feature of AD. While some degree of cerebral Ab angiopathy involving the leptomeninges and intraparenchymal vessels occurs in almost all cases of AD, the proportion of microvessels within a neocortical region containing deposits of the Ab peptide is not known (87). In addition, the mechanisms behind the effects of several vascular factors and peripheral vascular pathophysiology might promote the late-onset of AD (79,87-89). Apolipoprotein E (ApoE), a major risk factor for atherosclerosis (8,90,91) as well as AD (92), may be linked to AD via its effects on the vasculature (6,7,67,93).
Thomas and coworkers determined the percentage of cerebral microvessels in the temporal cortex and parahippocampal gyrus that were associated with the predominant Ab1-42 form of the Ab peptide in clinically and pathologically confirmed cases of AD (87). Surprisingly, double immunostaining methods found that at least 40% of the microvessels in the two brain regions contained Ab1-42 deposits (87). However, there was no correlation of such localization with the ApoE genotype, although E4 homozygotes revealed a greater Ab1-40 burden. Observations suggest that a high proportion of cortical microvessels are associated with Ab1-42, which may affect microvascular function (87). Moreover, higher levels of low density lipoproteins (LDL) and ApoB in total serum are associated with increased deposition of Ab in demented individuals with neuropathologically confirmed AD (94). These findings indicate a key role for vascular abnormalities in the pathogenesis of AD. Since chronic hypoxia/hypoperfusion, Ab depositions, and AD are all maladies with similarities to atherosclerosis, one would expect them to share risk factors (6,9,64). Likewise, one would also expect that the same preventive interventions would alleviate their symptoms (4,64,94).
Hyperlipoproteinemia is associated with the impairment of NO-mediated, endothelium-dependent dilation. Galle and coworkers (17) demonstrated that oxidized lipoprotein(a) impairs endothelium-dependent dilation and is more potent than oxidized LDL in this effect. Comparisons between ventricular fluid (VF) lipoproteins isolated from AD patients and non-demented age-matched patients show that cerebrospinal fluid (CSF) lipoprotein metabolism is altered in AD (95). These data support the hypothesis that there is a direct relationship between vascular and lipoprotein abnormalities in AD. The positive linear relationship between AD and fat intake is additionally relevant (31,96).
A recent study (97) showed direct evidence linking cholesterol metabolism and the development of AD in a transgenic mouse model. This work also indicated that diet-induced hypercholesterolemia results in significantly increased levels of formic acid-extractable Ab peptides in the central nervous system (CNS) of AD mice. The total level of Ab was strongly correlated with the level of cholesterol in both the plasma and CNS. The Ab level also correlated with the number and size of amyloid deposits (97). These data demonstrate that dietary cholesterol increases Ab accumulation and accelerates AD-related pathology in animals. In addition, the study demonstrated that the ultrastructural features of vascular lesions and mitochondria in brain vascular wall cells from biopsy, human short postmortem brain tissues, and transgenic mice overexpressing AbPP (yeast artificial chromosome (YAC R140) and C57B6/SJL transgenic positive (Tg+) mice) all have the same pattern (64).
In situ hybridization using probes for human normal, a 5kb-deleted fragment, and mouse mitochondrial DNA (mtDNA) as well as immunocytochemistry using antibodies against AbPP, 8-hydroxy-2’-guanosine (8OHG), cytochrome c oxidase subunit 1 (COX), and lipoic acid revealed similar patterns of ultrastructural localization (64,98). There was a higher degree of amyloid deposition in the vascular walls of human AD and YAC and C57B6/SJL Tg+ mice compared to aged-matched controls (64). In addition, vessels with more severe lesions showed immunopositive staining for AbPP and possessed large, lipid-laden vacuoles in the cytoplasm of ECs. Significantly more mitochondrial abnormalities were seen in human AD microvessels as well as YAC and C57B6/SJL Tg+ mouse microvessels where lesions occurred (64).
In situ hybridization using normal and chimera (5 kb) mtDNA probes revealed positive signals in severely damaged mitochondria located in the vascular endothelium and in perivascular cells of lesioned microvessels close to regions of Ab deposition. These features were absent in undamaged regions of human AD tissues, YAC and C57B6/SJL Tg+ mouse tissues, and in age-matched control subjects. Vessels with atherosclerotic lesions revealed endothelium and perivascular cells possessing clusters of normal and deleted mtDNA (64). These mtDNA deletions were accompanied by increased amounts of immunoreactive AbPP, 8OHG, and COX in the same cellular compartmen (64). The above correlative observations demonstrate that vascular wall cells, especially their mitochondria, appear to be central targets for oxidative stress-induced damage before the development of AD pathology (64). On the other hand, the positive correlation between AD and cholesterol levels suggests that antioxidant therapy and cholesterol lowering drugs could delay the occurrence of AD (99,100). However, despite their frequencies, the pathophysiological and morphological changes in brain microcirculation that accompany AD remain poorly understood, and the specific factor controlling vascular tone in AD is unknown.
The role of mitochondrial abnormalities in the pathogenesis of oxidative stress-induced brain lesions during the development of AD
In aerobic cells, 90-95% of the total amount of ATP production comes from aerobic metabolism. The synthesis of ATP via the mitochondrial respiratory chain is the result of electron transport coupled to oxidative phosphorylation (for review and ref. see (101). Excitotoxicity, mitochondrial dysfunction, and free radical-induced oxidative damage have all been implicated in the pathogenesis of several different neurodegenerative diseases in addition to AD, and include PD, amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD). The main radical produced by mitochondria is the superoxide anion. Intramitochondrial antioxidant systems scavenge this radical to avoid oxidative damage, which can lead to impaired ATP production (102-104). Both processes, i.e., defective ATP production and increased oxygen radicals, may induce mitochondrial-dependent cell death (for more information see the review by Schulz and colleagues (104). During aging and some neurodegenerative diseases, including AD, damaged mitochondria are unable to maintain the energy demands of the cell (105). This can lead to an increased production of free radicals, which induces the interruption of oxidative phosphorylation and results in decreased levels of ATP (104).
Much of the interest in the association of neurodegeneration with mitochondrial dysfunction and oxidative damage emerged from animal studies using mitochondrial toxins (104). These dysfunctions have been strongly implicated in the pathogenesis of human as well as in animal models of neurodegenerative diseases (106-109), particularly AD (64,65,102,103,105,110). The effect of acute ischemia and chronic neurodegenerative diseases on neuronal mitochondrial ultrastructure has been reviewed recently (103). After long-term ischemia/reperfusion the mitochondrial ultrastructure disintegrates in vivo and in vitro (8,51,52). Apoptosis of degenerating neurons occurs in association with the accumulation of perikaryal mitochondria and oxidative damage to the nucleus (111).
This same pattern of mitochondrial lesions is observed in brain biopsy samples of human AD cases (65,105). The reduced expression of both mitochondrial and nuclear DNA-encoded genes is consistent with a physiological down-regulation of the mitochondrial respiratory chain in response to declining neuronal activity (102,103,109,110,112). However, the role of somatic cells and mitochondrial DNA mutations in the pathogenesis of mitochondria failure during AD is still controversial (103,109,110). Our recent findings indicate that mitochondrial abnormalities appear to be key features in the development of AD-like pathology in YAC AbPP transgenic mice (64,113-115). In humans, deleted mtDNA is increased at least 3-fold in AD cases as compared to controls (105). Moreover, it has been reported that mitochondrial DNA isolated from the brains of AD patients shows oxidative modifications containing 8-hydroxy-2’-deoxyguanosine (8OHdG) (38-40).
Additionally, studies using in situ markers for 8OHdG and 8-hydroxy-guanosine (8OHG) showed that RNA oxidation is a prominent feature of damaged neurons in AD (41-43). Quantitative analysis revealed a strong positive correlation between mtDNA deletions and cytoplasmic RNA oxidation among age-matched controls (r = 0.934) and AD neurons in the early stages of nonreversible damage (that is, in neurons which still contained relatively intact cytoplasmic organelles). However, no correlation existed for AD neurons in the end stages of nonreversible cellular damage (105). This result is due to the fact that end-stage neurons contain only remnants of cytoplasmic organelles, and thus, they vary in their amount of mtDNA.
Subcellular mechanisms and ultrastructural changes in the development of human AD
Our research group has been able to demonstrate specific immunocytochemical and molecular biological assays of human AD and transgenic (Tg+) mice overexpressing AbPP and ApoE4 as a model for Alzheimer’s disease (2,23,64,116-121). We used the C57B6/SJL Tg+ mouse model of AD to assess the binding of basic fibroblast growth factor (bFGF) and serum amyloid P component (SAP) to Ab as a measure of BBB integrity (119). Adjacent sections of brain were either stained with 4G8, a monoclonal antibody to Ab, or with basic fibroblast growth factor (bFGF) binding followed by 48.1, a monoclonal antibody against bFGF, Figure 1. The bFGF bound specifically to Ab neuritic plaques and the basal membrane (BM) of cerebral microvessels, Figure 1 and 2. However, no SAP immunoreactivity was found in the Tg+ mouse brain, suggesting that the pathogenesis of BBB impairment in this mouse model differs from that of AD, Figure 2. Abnormal mitochondria and lipofuscin were characteristic features of damaged hippocampal neurons in aged Tg+ mice, suggesting a direct relationship between vascular abnormalities, BBB breakdown, neuronal loss, and amyloid depositions, Figure 13-16, microscopy (EM) and cytochemistry revealed different sizes and types of Ab deposits in brain tissues of YAC AbPP mice (Figs. 3-10).
The cortical neuronal cell bodies in YAC AbPP and C57B6/SJL(Tg+) mice, transgenic for human AbPP, were characterized by different degrees of ultrastructural alterations in their mitochondrial structures, as is seen in AD (Fig. 11). In situ hybridization analysis with mouse and human mtDNA probes found a substantial amount of 5kb-deleted mtDNA in human AD and YAC AbPP mice hippocampal cellular compartments compared to aged controls, Figure 19,20 (64,116,117,122). The majority of these mtDNA deletions were found in mitochondrial-derived lysosomes of neurons, vascular wall cells (Figs. 22-23), and glia in regions closely associated with lipofuscin. This evidence suggests that, at least within AbPP YAC mice, the proliferation, deletion, and duplication of mtDNA occurs most frequently in mitochondria that have been fused with lysosomes (64,116,117), Figure 22-23. Biopsy samples from human AD brains were dominated by abnormal mitochondria in comparison to a control group, Figure 11 and 21.
In situ hybridization with a chimeric cDNA probe for the 5kb common deletion indicated that the 5kb-mtDNA is increased at least threefold in AD neurons as compared to control cases, Figure 21. Using quantitative analysis, we found a strong positive correlation between mtDNA deletions and 8OHG in the same cases (r=0.934; Figure 17. Ultrastructural localization of mtDNA in situ hybridization with colloidal gold showed that deleted mtDNA is mainly found in abnormal mitochondria, Figure 20 and 21. Only hippocampal and cortical vulnerable neurons showed immunopositive staining for 8OHG in AD (64, 98,116,117,123,124) Figure 21.
In addition, the capillary EC and perivascular pericytes showed a high level of 8OHG immunostaining (64,116,117,122). This data strongly indicates that the oxidative stress markers seen in the AD brain selectively affect the population of vulnerable neurons, vascular EC, and perivascular cells, further implying that oxidative stress-induced hypoperfusion plays a key role in the pathogenesis of AD. We recently studied the effect of ApoE4 overexpression on cerebral blood flow (CBF) in an attempt to determine if ApoE4 acted as an initiator of brain hypoperfusion. The comparison of APOE4 transgenic mice with age-matched WT mice demonstrated that any neuronal, glial, and microvascular pathology is associated with a significantly increased percentage of abnormal compared to normal mitochondria, as well as with an increase in the total number of damaged mitochondria; that is, those mitochondria with electron-dense matrices, broken cristae, and mitochondria-derived lysosomes, Figure 22-23.
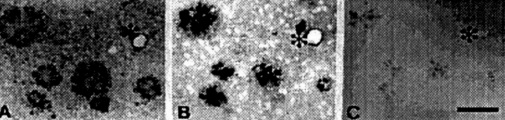
Figure 1. (above) Adjacent sections of transgenic mouse brain were stained with 4G8, a monoclonal antibody to amyloid (A), with bFGF binding followed by 48.1, a monoclonal antibody against bFGF (B), and with heparinase pretreatment before bFGF binding (C). The cores of amyloid plaques are intensely stained with 4G8, and bFGF binding is colocalized with amyloid immunoreactivity. bFGF also binds to the BM of cerebral microvessels walls (*). bFGF binding is greatly diminished by heparinase pretreatment (C).
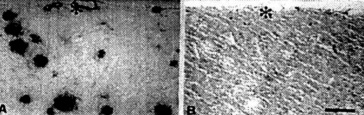
Figure 2. (above) Adjacent sections of transgenic mouse brain sections were immunostained with RPG9, a polyclonal antiserum to amyloid (A), and rabbit antiserum to serum amyloid protein (SAP) (B). The core of amyloid plaque and the BM of cerebral vessel wall (*) are intensely stained by RPG9. There is no evidence of immunoreactivity of SAP in amyloid plaques.
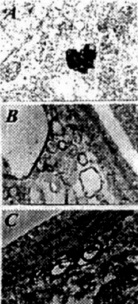
Figure 3. (left) C57B6/CJL transgenic mice show the different degrees of Ab immunostaining in the neuronal cell body (A) and vascular wall cells (B and C).
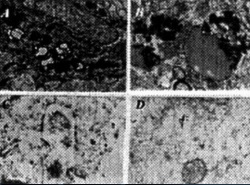
Figura 4. (left) The ltrastructural features of a bFGF stain from an aged C57B6/CJL transgenic mouse. (A) bFGF and (B) Ab PAP immunotechniques. Immunopositive products were seen in the cell bodies of damaged neurons. (C and D): Double immunogold labeling using the antibody against Ab and bFGl (10 and 17nm respectively).
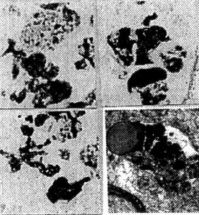
Figure 5. (above) YAC APP homozygous aged mice: Hippocampus. Amyloid immunostainin. using monoclonal antibody reveals different sizes of extracellular amyloid deposits. Thin sections without uranylacetate and lead citrate staining. The neuronal cell body also shows the presence of amyloid depositions.
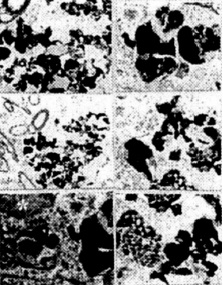
Figure 6. (left) The features of amyloid depositions (stained using a monoclonal antibody) in a neuronal eel body and the extracellular matrix in YAC APP age-matched transgenic mice. Amyloid depositions were associated with the formation of PHF-like structures (asterisk).
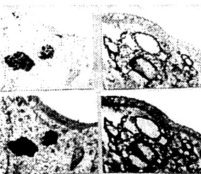
Figure 7. (left) The architecture of amyloid staining in the vascular wall of YAC APP transgenic mice. A and B: Without uranylacetate and lead citrate count staining. C and D: The same regions with count staining.
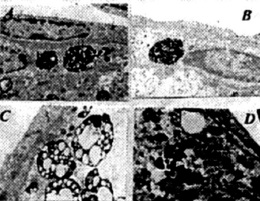
Figure 8. (above) The features of APP immunostaining in frontal cortical microvessels in YAC APP transgenic mice: A, B and C are without count staining. D is with count staining.
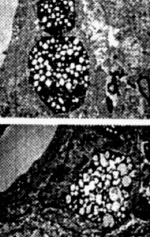
Figure 9. (left) Aged YAC APP transgenic mice microvessels show atherosclerotic changes. Large amounts of lipid vacuoles were seen in the matrix of the vascular endothelium and perivascular pericytes.
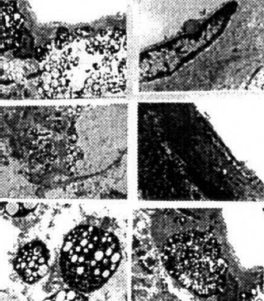
Figure 10. (above) The features of atherosclerotic like changes in brain microvessels of aged YAC APP transgenic mice. Lesions were heterogeneous and correlated with the degree of EC lesions.
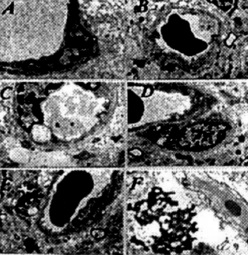
Figure 11. (above) The damage was seen in the vascular EC and perivascular cells in AD biopsy, but not in control brain (A-B). In addition, these vascular abnormalities were associated with amyloid depositions around the vessel wall (E-F).
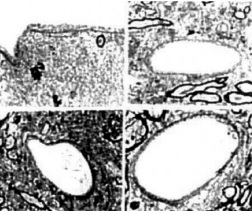
Figure 12. (above) Age matched control mice brain vessels did not show any particular lesions in the ultrastructure of vascular endothelium at different levels of microcirculation. Only sometimes a very small amount of lipid droplets appear in the matrix of pcrivascular cells.

Figure 13. (left) In age-matched YAC API1 uansgenic mice frontal coilical neurons, different stages of mitochondria lesions occurred in the neuronal cell body Giant and electron dense (ED) mitochondria appeared to be permanent features of the neuronal abnormality.
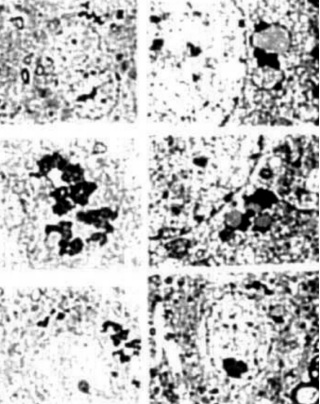
Figure 14. (rigth) APP immunostaining in cortical neurons of YAC APP transgenic mice. The APP immunopositive reaction was associated with the degree of mitochondria 745 abnormality and the presence of a giant size of lipofuscin in the neuronal cell body.
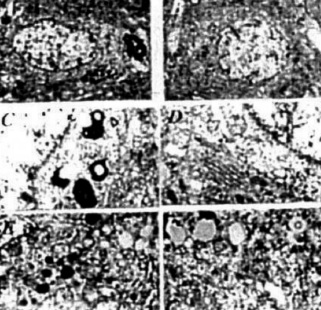
Figure 15. (left) AD brain biopsy Neurons with different grades of ultrastructural lesions under lower (A and B) and higher magnification (C-F) In the neuronal cell body mitochondria lesions were associated with lipfuscin formation and mitochondria appeared to be a major substrate for their formation. However, where microtubules were present the mitochondria did not show any particular ultrastructual changes.
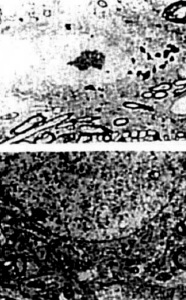
Figure 16. (rigth) Age-matched control, non transgenic mice did not show any particular changes in their neuronal ultrastruclure. In some neurons lipofuscin was sometimes seen. However, the mitochondria did not exhibit any particular changes in their ultrastructure.
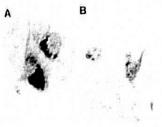
Figure 17. (left) Pyramidal itncurons of the hippocampus are ^highly vulnerable to death in AD, showing increased mtDNA with the 5kb common deletion in all cases of AD (A) as compared to controls (B)
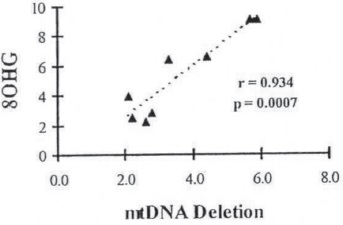
Figure 18. The extent of oxidativc damage (8OHG) is highly dependent on the degree of mt abnormalities, in arbitrary units.

Figure 19. mtDNA accumulates in abnormal mitochondria and is prominent in vesicles fused with lipofuscin. Colloidal gold (17nm) label denotes in situ hybridization for mtDNA with the common 5kb deletion.
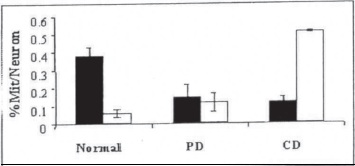
Figure 20. The percentage of different types of mitochondria with cristac (normal) and with partial (PD) or complete (CD) cristae destruction in brain biopsy samples from control (n = 6) and AD (n = 8) patients.
We theorize that vascular abnormalities, especially mitochondrial lesions and increased oxidative stress markers in the cellular and subcellular compartment, are responsible for altering regional blood flow, which can lead to BBB damage and breakage during the development of AD. Therefore, future studies examining the significance of mitochondrial pathophysiology in different cellular compartments may provide important insight into neurodegenerative disease pathobiology and provide targets for treating these conditions (2,23,120).
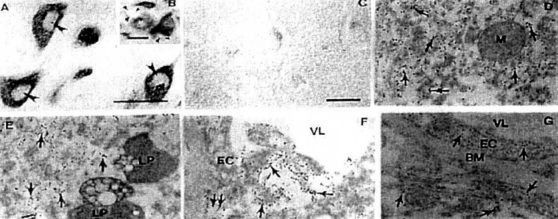
Figure 21. The vulnerable neurons (A-B) and vascular and non-vascular cells (F-G) show positive immunoreactivity for 8 OHG in AD, but not in age-matched control (C) Damaged mitochondria (double arrow) and cytoplasmic matrix (arrows) of neurons (D-E) and EC (F-G) also show the 8-OHG immunopositivc staining Lipofuscin (LP) and BM contained no 8-OHG.
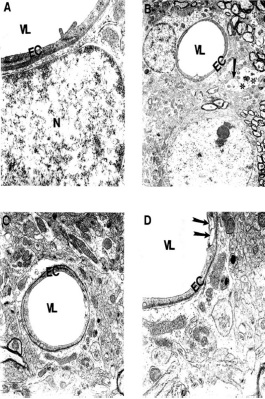
Figure 22. (above) Ultrastructural changes of cortical microvessels from young (A-B) and aged (C-D) ApoE4 Tg mice. Microvessels from young ApoE4 mice show the stress reaction of vascular endothelium. Destruction was also seen in the matrix of perivascular nerve terminals (arrow) and perivascular cells (indicated by double asterisk). Magnification: A) 25,000; B) 5,000; C) 10,000; D) 20,000. Reprinted with permission of J. Neurological Science, Shenk et al., 2008. (120).
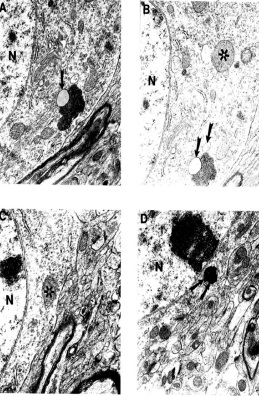
Figure 23. (above) Ultrastructural feature of age-associated neuronal mitochondrial change in ApoE4 Tg(+) mice. Mitochondria-derived lysosome association with lipofuscin appears to be the main feature of mitochondrial damage. Arrowhead: Mitochondria-derived lysosomes. Asterisk: Normal mitochondria. Double Arrowhead: Hypoxic (electron-dense) mitochondria. Magnification: A and B) 20,000; C) 25,000; D) 15,000. Reprinted with permission of J. Neurological Science, Shenk et al., 2008 (120).
Future studies must seek to answer the following questions: (1) What are the major factors altering and/or controlling cerebral blood flow during chronic hypoperfusion and/or the development of atherosclerotic changes in brain microvessels? (2) What are the roles of vasoactive substances, namely NO and ET-1, during the development of these changes? (3) Does chronic hypoperfusion with concomitant oxidative stress accelerate vascular and neuronal lesions, including mtDNA deletions, during normal aging and/or when the brain is exposed to chronic hypoxia? Resolving these issues will provide insight to develop novel therapeutic approaches that will modify the natural history of these chronic disorders associated with aging.