Bodas de plata de la reacción en cadena de la polimerasa (PCR)
Gladys Pinilla B1, Karen Cubillos2, Mónica Rodríguez13
1Universidad Colegio Mayor de Cundinamarca, Bogotá - Colombia.
2Instituto de Biotecnología Universidad Nacional, Bogotá - Colombia.
3Universidad Nacional Posgrado de Microbiología
Correspondencia: gpinillab@gmail.com
Recibido 27-04-08 / Aceptado 19-05-08
Palabras clave: agentes intercalantes, cuantificación absoluta, cuantificación relativa, curvas de calibración, gen normalizador, sondas fluorogénicas.
Truly revolutionary inventions have promoted the change of thought and the way to work in the laboratory. One of these inventions is the polymerase chain reaction (PCR), which has contributed significantly to the scientific knowledge. The different methodologies that use the polymerase chain reaction have allowed investigators to manipulate the genetic information of organisms, facilitating procedures like cloning and sequencing, among others, which sped the Human Genome Project results significantly. There are several variants of the conventional polymerase chain reaction. This work tries to present a revision on the subject, especially on the Real-time polymerase chain reaction, due to the advantages that it offers.
Key words: absolute quantification, calibration charts, fluorescent probe, normalizing gene, intercalate agents, relative quantification,
En 1989, Science selecciono la PCR como el principal desarrollo cientifico y la Taq polimerasa como la molecula del ano. El principio de la PCR consiste en determinar la secuencia de interes y seleccionar pequenos segmentos de nucleotidos llamados iniciadores o cebadores, complementarios con la secuencia de nucleotidos de los extremos opuestos de las cadenas que flanquean a dicha secuencia, a partir de los cuales mediante la accion de la Taq polimerasa, se inicia la elongacion o sintesis de nuevas cadenas en el extremo 3’ de cada iniciador, para obtener multiples copias de dicho segmento (2,3).
Las aplicaciones de la PCR son multiples, entre ellas la amplificacion de fragmentos de genes, modificacion de fragmentos de ADN, deteccion sensible de microorganismos y su genotipificacion, analisis de muestras arqueológicas y estudios antropologicos, la deteccion de mutaciones importantes en enfermedades hereditarias, transformación maligna o tipaje de tejidos, el analisis de marcadores geneticos para aplicaciones forenses, pruebas de paternidad y mapeo de rasgos hereditarios, estudio de expresion de genes y farmacogenomica, entre otras (4-6).
Variantes de la PCR convencional
Existe diversidad de variantes de la PCR convencional, algunas de las cuales se presentan en esta revision, sin embargo este articulo se enfoca en la PCR en tiempo real (PCRtr).
PCR inversa: se emplea para clonar regiones desconocidas de un ADN, situadas en posicion vecinal a secuencias diana conocidas. Es decir, en lugar de amplificar la region interna, flanqueada por los dos iniciadores (PCR convencional), se amplifica la region externa que flanquea los iniciadores. Para ello es necesario cortar el ADN a ambos lados de la region diana con una enzima de restriccion, de tal forma que los extremos cohesivos resultantes puedan hibridar entre si, formando una molecula circular. Esta es cerrada por una ligasa y se realiza la PCR con iniciadores que hibridan los extremos 5’ de la secuencia conocida, por lo que la elongacion se extendera alrededor del circulo generando copias de ADN delimitado (7).
PCR anidada (Nested-PCR): aumenta la especificidad al realizarse una segunda reaccion de PCR, con dos iniciadores nuevos que hibridan dentro del fragmento diana amplificado en la primera reaccion, permitiendo la obtencion de productos de PCR mas cortos y especificos (8).
PCR con adaptadores: permite amplificar una region de ADN de secuencia desconocida, ligando a fragmentos de restriccion, secuencias adaptadoras, es decir oligonucleotidos sinteticos con extremos cohesivos compatibles con los generados en la muestra. Los iniciadores son especificos para las secuencias 3’ de los adaptadores, permitiendo la amplificacion del conjunto de adaptadores y secuencia diana (8).
PCR asimétrica: se trata de generar copias de hebra sencilla de un ADN. Es la variante mas simple, en la que se anaden diferentes concentraciones de ambos iniciadores, de modo que tras los primeros ciclos de PCR uno de ellos se agota y deja disponible suficientes copias del ADN diana. Solo una de sus hebras sigue amplificando gracias al iniciador mas abundante (7).
PCR larga (L-PCR): su objetivo es superar los limites de la PCR convencional para amplificar con fidelidad regiones diana de gran tamano (entre 5 y 40kb), las cuales son muy convenientes para amplificar genomas virales, intrones, ADN mitocondrial y grupos de genes (8).
PCR múltiplex: las reacciones de PCR ocurren simultaneamente y en un mismo tubo, amplificando diferentes secuencias diana, para lo cual se usan varias parejas de iniciadores, permitiendo la deteccion e identificación simultanea de distintos genes de interes (9). RT-PCR (PCR con transcriptasa inversa): a partir de ARN diana y mediante la transcripcion reversa del ARN a ADNc, puede ser utilizado para la amplificacion, deteccion de la expresion de genes especificos o almacenaje de ADNc (7).
PCR en tiempo real
Generalidades
La cuantificacion de la expresion o cantidad de ARNADN presente en una muestra se realizo inicialmente mediante PCR competitiva. Pero fue desde finales de 1990 cuando la cuantificacion de la expresion logra importancia en la practica clinica; sin embargo, la PCRtr también permite la identificacion de nuevos genes y polimorfismos, el conocimiento de anomalias cromosomicas estructurales y epigeneticas (10).
La primera reaccion de PCR en tiempo real se llevo a cabo utilizando el marcador fluorescente bromuro de etidio, el cual aumenta su fluorescencia al unirse al ADN y la reaccion de PCR se evidencio con una videocamara. El primer equipamiento para realizar PCR en tiempo real fue comercializado en 1996 por Applied Biosystems.
Posteriormente se fueron incorporando distintas empresas como BioGene, Bioneer, Bio-Rad, Cepheid, Corbett Research, Idaho Technology, MJ Research, Roche, Applied Science, y Stratagene. En los equipos actuales, la deteccion del sistema incorpora modulos opticos que son capaces de detectar secuencias seleccionadas marcadas con fluorocromos (6,10,11,13).
En la PCRtr, los procesos de amplificacion y detección se producen de manera simultanea en el mismo vial cerrado, sin necesidad de ninguna accion posterior. Ademas mediante la deteccion por fluorescencia se puede medir durante la amplificacion la cantidad de ADN o
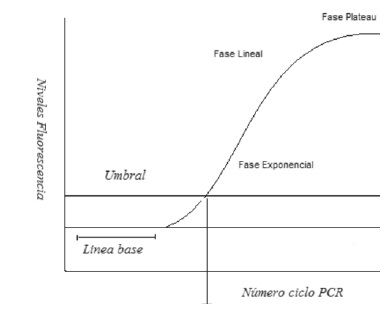
Figura 1. Cinética de la PCRtr.
ARN sintetizado en cada momento, ya que la emision de fluorescencia producida en la reaccion es proporcional a la cantidad de ADN o ARN formado, lo que permite conocer y registrar en todo momento la cinetica de la reaccion de amplificacion. La cuantificacion de la muestra por PCRtr incrementa la fiabilidad del analisis de la expresion del gen (12).
Cinética de la PCRtr
En la Figura 1 se observa la fase exponencial, fase lineal y fase Plateau. Ct, corresponde al inverso del log del número de copias. La linea de base es el nivel basal o background de fluorescencia durante los primeros ciclos de PCR. El umbral es el nivel de fluorescencia fijo por encima de la linea base.
En la fase exponencial temprana la fluorescencia supera el umbral. En la fase logarítmica lineal se duplica exactamente el producto en cada ciclo (optima amplificacion). Y en la fase de plateau, los reactivos se consumen, la reaccion se detiene (reacción de punto final).
La cinetica de amplificacion de PCRtr evidencia la acumulacion de la emision de fluorescencia en cada ciclo de reaccion, y esta puede ser clasificada en cuatro fases. En esta cinetica es importante resaltar el Umbral o Threshold (nivel de fluorescencia fijo por encima de la linea base) y el Ct o Cp: (ciclo del umbral o punto de cruce o corte), ciclo en el cual la fluorescencia supera el umbral y es detectada.
El Ct es inversamente proporcional a la concentración inicial de ADN o ARN diana presente en la misma; los datos que se obtienen permiten calcular la eficiencia de la amplificacion (14-17).
.
Eficiencia de la PCRtr
Es la proporcion a la que un amplicon de PCR se genera, normalmente expresado con un valor en porcentaje. Si un amplicon dobla la cantidad durante la fase logaritmica de su amplificacion por PCR, el ensayo tiene 100% eficiencia (18). La curva estandar de la PCRtr es representada graficamente como el semi-log del valor de CT versus el log del ácido nucleico inicial; una pendiente de la curva estandar de -3.32 indica una reaccion de PCR con 100% eficiencia, mientras que pendientes más negativas que -3.32 (ej. -3.9) indica reacciones que tienen menos del 100% de eficiencia, Figura 2. Pendientes mas positivas que –3.32 puede indicar problemas de calidad de la muestra o problemas de pipeteo.
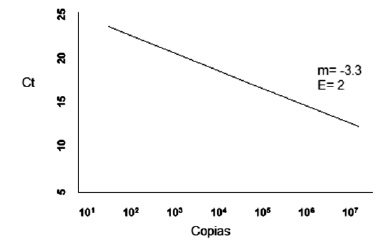
Figura 2. Gráfica eficiencia.
E= 10 -1/m
y= mx+b
Pendiente aceptable entre -3.2 y -3.8
Eficiencias entre: 83 y 105% =2 (Doble de copias)
La eficiencia de la PCRtr puede ser estimada mediante curvas de calibracion realizadas con diluciones seriadas de un estandar que puede ser ADN o un plasmido (19). Los parametros que afectan la eficiencia de PCR son: especificidad de primers y sondas, concentraciones altas de dNTPs (20), agentes inhibidores como hemoglobina, heparina, IgG, altas concentraciones de proteina (21,22).
Agentes intercalantes
Los sistemas de deteccion por fluorescencia empleados en la PCRtr pueden ser agentes intercalantes y sondas especificas marcadas con fluorocromos (13). Los agentes intercalantes son fluorocromos que aumentan notablemente la emision de fluorescencia cuando se unen al ADN de doble helice. El mas empleado es el SYBR Green I. El incremento del ADN en cada ciclo se refleja en un aumento proporcional de la fluorescencia emitida. Este sistema de deteccion tiene la ventaja de que la optimizacion de las condiciones de la reaccion es facil y mas economica que las sondas especificas. Para mejorar la especificidad se deben emplear condiciones de reacciones optimas y una seleccion cuidadosa de los primers para disminuir el riesgo de la formacion de dimeros; ademas, es recomendable iniciar la reaccion de sintesis de ADN a temperaturas elevadas (hot-start PCR).
La mayoria de los equipos para PCR en tiempo real tienen la posibilidad de determinar la temperatura de fusión de los fragmentos amplificados (Tm= temperatura a la que el 50% de la molecula de ADN esta desnaturalizado). Cada fragmento amplificado tiene una Tm caracteristica, que depende sobre todo de su longitud y de la composición de sus bases; esta aplicacion permite comprobar, la especificidad de los fragmentos detectados en la PCR (16,23).
Ventajas SYBR Green
- Diez veces más sensible que el bromuro de etidio.
- Más económico que el uso de sondas.
- Una sóla química para todos los experimentos.
- No requiere diseño de sondas.
Desventajas SYBR Green
- Requiere ajustar de manera más exhaustiva las condiciones.
- Difícil genotipificación de SNPs.
- No permite hacer PCR multiplex.
- Baja especificidad, debido a que se unen de manera indistinta a productos generados.
Análisis de curvas de disociación
Al utilizar como agente quimico para la PCRtr el SYBR Green, se debe realizar la curva de disociacion, para comprobar la especificidad de los fragmentos detectados en la PCR.
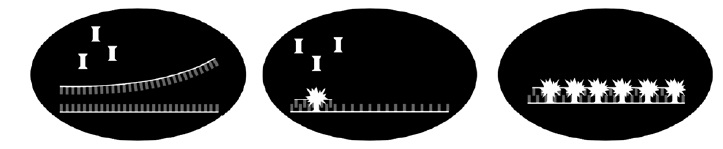
Figura 3. SYBR Green. Acción intercalante a lo largo de la cadena de DNA emitiendo fluorescencia
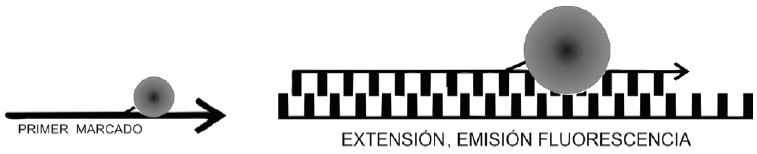
Figura 4. Efecto LU X (light upon extensión). El iniciador marcado con el fluoroforo emite fluorescencia en el momento que se hibrida con su blanco.

Figura 5. Sondas Hibridación: A. Sonda Taqman, B. Molecular Beacons, C: Iniciador escorpión y D: De hibridación. Sondas de PCR tiempo real que funcionan bajo el principio FRET, transferencia de energía fluorescente mediante resonancia
La curva de disociacion se basa en un gradiente de temperaturas crecientes despues de la PCRtr, para monitorizar la cinetica de disociacion de los fragmentos amplificados y comprobar su especificidad. Es decir, se detecta la amplificacion del blanco ADN o ARN (primera derivada) y dimeros de primers (segunda derivada).
Iniciadores marcados con fluoroforos
“Los primers LUX fluorogenic” son un nuevo producto que permiten la deteccion de la secuencia blanco por emision de fluorescencia. LUX (light upon extension) es un iniciador especifico a la secuencia de interes que se encuentra marcado con un fluoroforo, cuando el iniciador se hibrida con la cadena blanco, el fluoroforo es excitado, resultando en incremento significativo de senal de fluorescencia, Figura 4.
Este producto tiene como ventaja que es menos costoso que las sondas y mas sensible que el SYBR Green (17,24).
Sondas de hibridación
Son sondas marcadas con dos tipos de fluorocromos, un donador y un aceptor. El proceso se basa en la transferencia de energia fluorescente mediante resonancia (FRET) entre las dos moleculas. Este principio consiste en que una molecula de alta energia cercana a una molecula de baja energia (quencher), promueve una transferencia energetica y no habra emision de fluorescencia.
Una vez se separan dichas moleculas se emite la fluorescencia que es captada por el lector del equipo (25).
Entre las sondas se encuentran:
Sondas TaqMan: sonda corta que necesita la ruptura de la polimerasa para separar las dos moleculas fluorecentes y liberar fluorescencia, Figura 5A (26).
Molecular Beacons: sonda cerrada que hibrida con su blanco durante la PCR liberando fluorescencia, Figura 5B (4).
Iniciador escorpión: incluye un iniciador de PCR, que al unirse con una region especifica del ADN blanco, el oligonucleotido unido al quencher se libera, plegandose la sonda marcada con el fluorocromo sobre el acido nucleico blanco liberando la fluorescencia, Figura 5C.
Sondas de hibridación: conformada por dos sondas. Si esta hibridada no emite fluorescencia a temperatura de 55°C, se libera la fluorescencia en fase de desnaturalización a 95°C, Figura 5D (6,17).
Controles para PCR
Control Interno: se define como una secuencia de ADN o ADNc que es co-amplificado con el ADN blanco y se usa principalmente para aumentar la confiabilidad de los ensayos, ya que permite la deteccion de falsos negativos.
Es decir determina un ensayo fallido o inhibicion de la reaccion. El control interno mas usado para la PCRtr es un plasmido que contiene secuencias de organismos distantes filogeneticamente. Estos plasmidos pueden ser adicionados a la mezcla de PCR o directamente a la muestra clínica (de esta forma se verificaria si la extraccion de ADN a partir de la muestra se llevo a cabo correctamente). La longitud del templado del control interno debe ser mayor a la longitud del templado de ADN blanco para asegurar que la competencia de la reaccion tienda mas hacia el ultimo (27).
Control positivo ó externo: no permite verificar la reaccion individualmente ya que la reaccion se da en un tubo diferente; no revela la ineficiencia en la extracción de ADN o ARN.
Control negativo: se puede utilizar otro organismo no relacionado con el organismo en estudio, o tambien se puede utilizar agua o buffer en lugar de ADN templete. Este control puede ser utilizado a la vez como control ambiental (28,29).
Tipos de detección
1. Cualitativa: presencia de secuencia blanco en la muestra.
2. Cuantitativa:
- Absoluta: cantidad moleculas en valor absoluto.
*Curva estándar.
- Relativa: relacion entre la expresion del gen blanco y un gen de referencia.
* Se requiere hallar el ΔΔCT (ΔCT del gen blanco y ΔCT del gen de referencia)(30).
* Método PFaffl.
Cuantificación absoluta
En la cuantificacion absoluta se realiza la comparación con amplificados de los cuales se conoce el numero absoluto de moleculas iniciales a cuantificar (estandares), se amplifican en una misma reaccion con la muestras de interes, pero en tubos diferentes. La curva estandar utiliza diluciones seriadas (minimo 5) de estandares de concentraciones conocidas; el número de moléculas es calculado como una extrapolación a partir de una curva generada por los estandares. Este metodo asume que todos los estandares y las muestras tienen aproximadamente igual eficiencia de amplificacion.
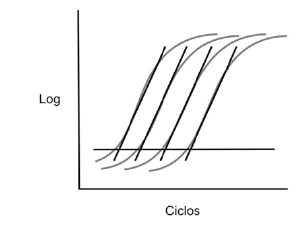
Figura 6. Curva estándar para cuantificación absoluta.
Se obtiene el valor absoluto como numero de copias, celulas, μg/μl, etc., Figura 6 (17,30,31).
Cuantificación relativa
Permite cuantificar las diferencias en la expresión del gen blanco relativo a la expresión del gen normalizador (30). La cuantificación relativa es util y sencilla para analizar la expresión entre genes (31,32).

Método de Ct comparativo (Método ΔΔCT)
El método de CT comparativo, también llamado el método de ΔΔCT, es similar al método de la curva estándar relativa, excepto que este usa las formulas aritméticas para lograr un resultado para la cuantificación relativa.
Es posible eliminar el uso de curvas estándar y usar el método de ΔΔCT, pero las eficacias de PCR entre el blanco (s) y control (s) (endogeno) deben ser relativamente equivalentes (16). La ventaja de este metodo es que no exige curvas estándar para cada pozo de reacción, permitiendo el ahorro de reactivos y es útil cuando hay un número alto de blancos y/o de muestras (33).
Normalizadores
Los normalizadores son un grupo de genes que se expresan en todas las celulas del organismo y codifican para proteínas que son esenciales para el funcionamiento general de las celulas; pueden ser expresados en diferentes condiciones experimentales incluyendo diferentes tejidos o tipo de celulas.
Su expresion no debe ser muy diferente del gen blanco y la estabilidad y tamano debe ser parecido a los genes de interes. Los datos de la expresion de genes normalizadores son utilizados para corregir la variacion muestra-muestra y compensar el error experimental (28,29).
Recientes estudios muestran que la expresion de muchos genes, que son usados como normalizadores en experimentos en PCRrt, pueden ser influenciados por la actividad metabolica, por la fase de crecimiento y por las condiciones experimentales. Por lo cual la normalización con un unico gen “Housekeepin”puede falsear los resultados. Recientes estudios proponen el uso de dos o mas genes normalizadores (16).
Genes que se han estudiado durante crecimiento in vitro y bajo diferentes condiciones para determinar cuales pueden ser utilizados como normalizadores para la expresion de genes bacterianos son:
Gen 16S rRNA: (1500pb) parte esencial del complejo ribosomal. La concentracion celular de ribosomas es proporcional a la sintesis de proteina total y de la actividad metabolica celular. Por lo cual es un buen marcador del contenido ribosomal, cuyos transcriptos tienen una estabilidad excepcional, ya que el 90% de todos ellos, durante la fase logaritmica, tienen una vida media de 5 minutos y estabilidad hasta por 3 horas (34,35,36).
Gen gmk: (2937 pb), codifica una enzima (guanilato kinasa) esencial para la sintesis de guanosina. Es un componente del metabolismo celular bien conocido, que puede variar considerablemente bajo diferentes condiciones (35,37,38).
Gen tpi: (3373 pb), representa un gen Houeskeeping vital, su producto (triosefosfato isomerasa) es esencial durante la glicolisis. Las secuencias que codifica este gen tanto en eucariotas como en procariotas son altamente conservadas entre especies filogeneticamente distintas (39,40).
Gen dfhr: (3309pb) su producto, la dihidrofolato reductasa esencial para la sintesis del acido folico; es clave en el camino del tetrahidrofolato y esencial en la biosíntesis de las purinas, timidilato y varios aminoacidos (35).
Gen hsp-60: esta involucrado en el plegamiento y ensamblaje de proteinas y en la reactivacion de proteínas denaturadas (35).
Gen fabD: codifica para una proteina presente en todas las bacterias, la malonil CoA-ACP transacetilasa (MCAT). Esta enzima, esencial para la biosintesis de acidos grasos (sustrato para la elongacion durante la sintesis de acidos grasos) y por tanto importante en la sintesis de los acidos
micolicos (35).
Entre otros normalizadores estan el gen glyA, gyrA, pyk, recF, rho, rpoD, rrsC (41-43).
Como normalizadores para la expresion de genes humanos se han usado:
rRNA ribosomal 28S: es uno de los mas usados, que ha mostrado la mayor estabilidad en una gran variedad de tejidos. No es tan conservado en diferentes especies; puede permanecer intacto en caso de degradacion, constituye mas del 50% del RNA total y su expresion se afecta por procesos celulares y drogas (44).
Actina: uno de los mayores componentes de los microfilamentos del citoplasma en eucariotas, tiene diferentes papeles en funciones celulares, principalmente en estructura y morfologia celular. Su expresion se ha visto alterada en tejidos en diferentes estados de desarrollo, diferentes cultivos de tejidos, bajo la infección de patogenos. En general no se recomienda su uso en condiciones que afecten la morfologia celular (44).
GAPDH: (Gliceraldehido 3-fosfato deshidrogenasa) es una enzima multifuncional involucrada en metabolismo celular. Al ser un gen de bajo numero de copias tiene una baja frecuencia de isoformas o pseudogenes lo que disminuye la posibilidad de reaccion cruzada. Su expresion se ha visto alterada en hipoxia, lineas celulares cancerigenas, en ontogenia y pancreatitis aguda (25).
Ciclofilina: tiene funcion en la catalisis del doblamiento de proteinas por rotacion del enlace peptidico en los enlaces de serinas, actua como chaperona en el trafico de proteinas asi como en la degradacion nucleolitica del genoma. Su expresion se ha visto alterada en diferentes etapas de desarrollo y bajo ciertos tipos de estres como choque termico, infeccion viral, y en exposicion ante el acido salicilico (45).
EF-1a: (Elongation Factor 1a) es una proteina ubiquita que une los tARN a los ribosomas durante la sintesis de proteinas. Al ser parte del sistema de traduccion, su expresion se puede ver afectada en tejidos de rapido crecimiento y desarrollo como meristemos vegetales. Se ve afectada tambien en situaciones que alteran el crecimiento y desarrollo celular como muerte y envejecimiento celular (45-47).
Evaluación de la expresión génica
Para la evaluacion de la expresion genica mediante los metodos clasicos se requiere de una gran cantidad de ARN mensajero que es dificil de obtener cuando el numero de muestras es limitado o cuando el material biologico es una poblacion de diferentes tipos (25). Esta tecnica ha sido usada para la cuantificacion de ARN de diversas células como es el caso de la expresion genica de los ovocitos.
Los estudios sobre la oogenesis son escasos, dado que la diferenciacion se lleva a cabo en estadios fetales, por lo que son inaccesibles a la manipulacion experimental, y la cantidad de material biologico disponible para su analisis es escaso, Por lo tanto esta tecnica abre las posibilidades para ser utilizada en el analisis molecular de celulas obtenidas de cultivos biologicos o mediante microdiseccion con laser (48).
Diagnóstico microbiológico en alimentos
El diagnostico automatizado de patogenos en alimentos, continua siendo una necesidad para la industria y la salud publica, debido a esto la PCR se convirtio en una herramienta poderosa para el diagnostico microbiologico, pero debe cumplir optimamente muchos criterios analiticos, alta probabilidad de deteccion, baja contaminacion y protocolos comodos para la aplicacion y la interpretacion.
La segunda generacion de la PCR, es decir en tiempo real, tiene la precision y el potencial para la deteccion en un solo paso; ademas la cuantificacion del patogeno no se basa en un punto final sino en un incremento exponencial de la cantidad de ADN inicial (49).
La PCRtr esta siendo implementada para detectar microorganismos patogenos en alimentos, en muestras ambientales y en muestras clinicas. El diagnostico molecular de salmonelosis no tifoidea, cada vez se pone mas en auge, debido a la alta incidencia de intoxicación alimentaria asociada con la contaminacion de alimentos de origen animal como huevos y la carne de pollo (49,50).
La PCRtr es una herramienta fiable y rapida para el monitoreo de las muestras alimenticias a lo largo de la cadena de produccion ya que en tan solo 24 horas se detectan celulas en suspension de 103UFC/ml con una probabilidad del 70% o 104UFC/ml con una probabilidad del 100% (50,51, 52).
La bacteria Clostridium botulinum, es la responsable del botulismo, enfermedad neuroparalitica. Siete tipos de toxinas antigenicas han sido identificadas (A-G) BoNT (neurotoxina botulinica) tipo A, B E y raramente F son las responsables de botulismo humano (53). Esta bacteria puede contaminar alimentos mal enlatados o almacenados en recipientes abiertos o inapropiados.
Los métodos microbiologicos estandar toman solo en consideración la deteccion de C. botulinum; el bioensayo en ratones es el metodo universal, altamente sensible, pero costoso y con problemas eticos ya que requiere gran cantidad de animales. La deteccion de genes de BoNT por PCRtr es mas sensible y una alternativa mas rapida que los bioensayos (53, 54).
Diagnostico microbiológico clínico
El descubrimiento temprano y el tratamiento adecuado para la infeccion bacteriana tienen un gran impacto ya que en la mayoria de las infecciones se tratan empíricamente con antibioticos de amplio espectro debido al tiempo que se requiere, de 24 a 48 h para el procesamiento microbiologico de rutina (55).
Actualmente en pacientes en cuidado critico, que requieren la deteccion temprana de ADN bacteriano en el torrente circulatorio, es posible analizar las muestras clínicas como sangre (56), plasma (57), fluido cerebroespinal (58, 59) y otros especimenes mediante PCRtr (59,60). En contraste con estos nuevos acercamientos de diagnostico, los cultivos de sangre requieren mas tiempo y a menudo brindan resultados falsos negativos, debido a baja sensibilidad (61).
La PCRtr, se ha usado para la identificacion de mutaciones puntuales asociadas con resistencias a farmacos antimicrobianos (62). En solo una hora se puede determinar la presencia en heces de Enterococos resistentes a vancomicina (18), facilitando el control de la transmisión de este patogeno.
Tambien se ha empleado en la detección de mutaciones asociadas con resistencias a meticilina (31, 63) en S. aureus, a rifampicina y a isoniacida en Mycobacterium tuberculosis (18), y de mutaciones asociadas con resistencia a agentes antiviricos, como la lamivudina en VHB (41). Existe gran variedad de enfermedades ocasionadas por microorganismos que requieren diagnostico temprano.
Dentro de estas, la neumonia adquirida en la comunidad o nosocomial causada por la Legionella pneumophila, que se caracteriza por su crecimiento lento, por requerir medios de cultivo selectivos y periodos prolongados de incubacion. Para su diagnostico se usa generalmente metodos serologicos, sin embargo la deteccion de IgG e IgM no permite el diagnostico temprano. Se ha probado la deteccion de antigeno urinario y las amplificaciones de acido nucleico en muestras respiratorias (64). La PCRtr ofrece ventajas como alta sensibilidad, obtencion rapida de resultados y el potencial de detectar infecciones causadas por varios serogrupos de Legionella pneumophila y no -pneumophila (65,66).
Chlamydophila pneumoniae y Mycoplasma pneumoniae son agentes etiologicos de neumonia. C. pneumoniae causa del 9-15% de los casos de neumonia y el M. pneumoniae del 7-22% (67, 68). La incidencia mas alta de Chlamydophila pneumoniae y Mycoplasma pneumoniae es en ninos de edad escolar, entre 5 a 14 anos de edad.
Los agentes causales de infecciones respiratorias son dificiles de distinguir. La PCRtr duplex o multiplex ha sido una gran herramienta para el diagnostico acertado en estas enfermedades, diferenciando en un mismo tubo agentes etiologicos diferentes como en el caso de Chlamydophila pneumoniae, Mycoplasma pneumoniae y Legionella pneumophila (69,70)
Otra aplicacion en investigacion basica con células bacterianas, ha sido la cuantificacion de ARN y la valuacion de su expresion. Este es el caso del estudio de transcriptos del gen mecA en Staphylococcus aureus meticilin resistente, en donde se correlaciona la presencia del gen, con su expresion y el efecto de sus genes represores e inductores sobre esta (62). A manera de cierre, la PCRtr continuara evolucionando, al ser una herramienta versatil, que permite su uso en gran cantidad de areas de investigacion basica y aplicada.
2. McPherson JD, Marra M, Hillier L, Waterston RH, Chinwalla A, Wallis J, et al. A physical map of the human genome. Nature. 200;409:934-941.
3. Rodriguez IP, Barrera H. La reaccion en cadena de la polimerasa a dos decadas de su invencion. Ciencia UANL.2004;7:323-332.
4. Mocellin S, Rossi CR, Pilati P, Nitti D,Marincola FM. Quantitative real-time PCR: a powerful ally in cancer research. Trends Mol Med. 2003;9:189-195.
5. Templeton KE, Scheltinga SA, Graffelman WA, Van Schie JM, Crielaard JW, Sillekens P, et al. Comparison and evaluation of realtime PCR, real-time nucleic acid sequence-based amplification, conventional PCR, and serology for diagnosis of Mycoplasma pneumoniae. J Clin Microbiol. 2003;41:4366–4371.
6. Costa J. Reaccion en cadena de la polimerasa (PCR) a tiempo real. Enferm Infecc Microbiol Clin. 2004;22:299-305.
7. Luque JC, Herraez SA. Texto ilustrado de Biologia Molecular e Ingenieria Genetica. Ed. Harcourt.2000. pg 313.
8. Innia M, Gelfand D, Sninsky J. PCR strategies. Academic Press New York USA.1995:325.
9. Choi S, Kim S-H, Kim H-J, Lee D-G, Choi J-H, Yoo J-H, et al. Multiplex PCR for the detection of genes encoding aminoglycoside modifying enzymes and methicillin resistance among Staphylococcus species. J. Korean Med Sci. 2003;18:631-636.
10. Jimenez A, Gomez R, Agirre X, Barrios M, Navarro G, Eneriz E, et al. PCR en tiempo real, una nueva herramienta para la toma de decisiones clinicas. Haematologica. 2006;91:27-34.
11. Kaltenboeck B, Wang C. Advances in real-time PCR: application to clinical laboratory diagnostics. Adv Clin Chem. 2005;40:219-259.
12. Heid CA, Stevens J, Livak JK, Williams M. Real Time Quantitative. Genome Res.1996;6:986-994.
13. Equipos para RT-PCR. Disponible en www.Biorad.com.
14. Rutledge RG, Cote C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res. 2003;31:3-6.
15. Bustin SA, Mueller R. Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin Sci.2005;109:365–379.
16. Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonak J, Lind K, et al. The real-time polymerase chain reaction. Mol Aspects Med. 2006;27:95–125.
17. Wong M, Medrano JF. Real-time PCR for mRNA quantitation. Biotechniques. 2005;39:75-85.
18. Peirson S, Butler J, Foster R. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003;31:e73.
19. Pfaffl M. A new mathematical model for relative quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001;29:e45.
20. Kainz P. The PCR plateau phase - towards an understanding of its limitations. Biochim Biophys Acta. 2000;1494:23-27.
21. Al-Soud WA, Jhonsson LJ, Radstrom P. Identification and Characterization of Immunoglobulin G in Blood as a Major Inhibitor of Diagnostic PCR. J Clin Microbiol. 2000;38:345-350.
22. Al-Soud WA, Radstrom P. Purification and characterization of PCR-inhibitory components in blood cells. J Clin Microbiol. 2001;39:485-493.
23. Gudnason H, Dufva M, Bang DD, Wolff A. Comparison of multiple DNA dyes for real-time PCR: effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res. 2007;35:1-8.
24. Nazarenko I, Lowe B, Darfler M, Ikonomi P, Schuster D, Rashtchian A. Multiplex quantitative PCR using self-quenched primers labeled with a single fluorophore. Nucleic Acids Res. 2002;30:e37.
25. Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol. 2002;29:23–39.
26. Provenzano M, Rossi C, Mocellin S. The usefulness of quantitative real-time PCR in immunogenetics. Third Quarter. 2001:89-91.
27. Burggraf S, Olgemoller B. Simple technique for internal control of real-time amplification assays. Clin Chem. 2004;50:819–825.
28. Memorias Curso teorico-practico de PCR en tiempo real. Corpogen. Julio 26 y 27 de 2007.
29. Espy MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, et al. Real-time PCR in clinical microbiology:applications for routine laboratory testing. Clin Microbiol Rev. 2006;19:165-256.
30. Yuan J, Reed A, Chen F, Stewart N. Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;85:1471-2105.
31. Pfaffl M, Hageleit M. Validities of mRNA quantification using recombinant RNA and recombinant DNA external calibration curves in real-time RT-PCR. Biotechnol Lett. 2001;23:275–282.
32. Gibson U, Heid CA, Williams PM. A Novel method for real time quantitative RT-PCR. Genome Res.1996;6:995-1001.
33. Livak K, Schmittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:1402–1408.
34. Zucol F, Ammann RA, Berger C, Aebi C, Altwegg M, Niggli FK, et al. Real-time quantitative broad range PCR assay for detection of the 16S rRNA gene followed by sequencing for species identification. J Clin Microbiol. 2006;44:2750-2759.
35. Vandecasteele SJ, Peetermans WE, Merckx R, Eldere V. Quantification of expression of Staphylococcus epidermidis housekeeping genes with Taqman quantitative PCR during in vitro growth and under different conditions. J Bacteriol. 2001;183:7094–7101.
36. Rodicio M, Mendoza M. Identificacion bacteriana mediante secuenciacion del ARNr 16S:fundamento,metodologia y aplicaciones en microbiologia clinica. Enferm Infecc Microbiol Clin. 2004;22:238-245.
37. Beck B, Hoelsmeyer M, Paul S, Downs D. A mutation in the essential gene gmk (encoding guanlyate kinase) generates a requirement for adenine at low temperature in Salmonella enterica. J Bacteriol. 2003;185:6732-6735.
38. Skurray R, Brow M. Identification of Suitable internal controls to study expression of a Staphylococcus aureus multidrug resistance systems by quantitative real-time PCR. J Microbiol Meth.2007;70:355-362.
39. Unkles S, Logsdon J, Robinson K, Kinghorn J, Duncan J. The tpiA gene is a transcripcional isomerase and glyceraldehyde-3-phosphate dehydrogenase in oomycota. J Bacteriol.1997;179:6816-6823.
40. Xu Y, Hong Y, Hall T. Rice triosephosphate isomerase gene 5’ sequence directs β-glucuronidase activity in transgenic tobacco but requires an intron for expression in rice. Plant Physiol.1994;106:459-467.
41 Plamannt D. Michael, Stauffer VG. Regulation of the Escherichia coli glyA Gene by the metR Gene Product and Homocysteine. J Bacteriol. 1989;171:4958-4962.
42. Purvis IJ, Loughlin L, Bettany AJ, Brown AJ. Translation and stability of an Escherichia coli beta galactosidase mRNA expressed under the control of pyruvate kinase sequences in Saccharomyces cerevisiae. Nucleic Acids Res. 1987;15:7963-7974.
43. Poysti J, Nathan, Oresnik J. Characterization of Sinorhizobium meliloti triose phosphate isomerase genes. J Bacteriol. 2007;189:3445–3451.
44. Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods. 2000;46:69-81.
45. Rubie C, Kempf K, Hans J, Su T, Tilton B, Georg T, et al. Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues. Mol Cell Probes.2005;19:101–109.
46. Dheda K, Huggett J, Bustin S, Johnson M, Rook G, Zumla A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 2004;37:112-119.
47. Huggett J, Dheda K, Bustin S, Zumla A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005;6:279–284.
48. Bonilla E, Parraga M, Lopez LA, Escolar F, Del Marzo J. Cuantificacion de la expresion genica a partir de un numero limitado de celulas mediante RT-PCR en tiempo real. Bioquimia. 2002;27:3-7.
49. Burkhard M, Charlotta L, Wagner M, Kra¨mer N, Hoorfar J. Enumeration of Salmonella bacteria in food and feed samples by real-time pcr for quantitative microbial risk assessment. Appl Environ Microbiol. 2008;74:1299-1304.
50. De Medici D, Croci L, Delibato E, Di Pasquale S, Filetici E, Toti L. Evaluation of DNA extraction methods for use in combination with SYBR Green I Real-Time PCR to detect Salmonella enterica serotype Enteritidis in Poultry. Appl Environ Microbiol. 2003;69:3456-3461.
51. Burkhard M, Paccassoni E, Fach P, Bunge C, Martin A, Helmuth R. Diagnostic Real-Time PCR for detection of Salmonella in food. Appl Environ Microbiol. 2004;70:7046-7052.
52. Artin I, Bjorkman P, Cronqvist J, Radstrom P, Holst E. First case of type E wound botulism diagnosed using real-time PCR. J Clin Microbiol. 2007;45:3589–3594.
53. Fenicia L, Anniballi F, De Medici D, Delibato E, Aureli P. SYBR green real-time PCR method to detect Clostridium botulinum type A. Appl Environ Microbiol. 2007;73:2891–2896.
54. Yoon SY, Chung GT, Kang DH, Ryu C, Yoo CK, Seong WK. Application of real-time pcr for quantitative detection of Clostridium botulinum Type A toxin gene in food. Microbiol Immunol. 2005;49:505 511.
55. Edwards KJ, Kaufmann ME, Unders NA. Rapid and accurate identification of coagulase-negative staphylococci by real-time PCR.. J Clin Microbiol. 2001;39:3047-3051.
56. Kane TD, Alexander JW, Johannigman JA. The detection of microbial DNA in the blood: a sensitive method for diagnosing bacteremia and/or bacterial translocation in surgical patients. Ann. Surg. 1998;227:1–9.
57. Cursons RT, Jeyerajah E, Sleigh JW. The use of polymerase chain reaction to detect septicemia in critically ill patients. Crit Care Med.1999;27:937–940.
58. Lu JJ, Perng CL, Lee SY, Wan CC. Use of PCR with universal primers and restriction endonuclease digestions for detection and identification of common bacterial pathogens in cerebrospinal fluid. J Clin Microbiol. 2000;38:2076–2080.
59. Rantakokko-Jalava K, Nikkari S, Jalava J, Eerola E, Skurnik M, Meurman O, et al. Direct amplification of rRNA genes in diagnosis of bacterial infections. J Clin Microbiol. 2000;38:32-39.
60. Mariani BD, Martin DS, Levine MJ, Booth RE Jr, Tuan RS. The coventry award polymerase chain reaction detection of bacterial infection in total knee arthroplasty. Clin Orthop Relat Res. 1996;331:11- 22.
61. Klaschik S, Lehmann LE, Raadts A, Book M, Hoeft A, Stuber F. Real-time PCR for detection and differentiation of gram-positive and gram-negative bacteria. J Clin Microbiol. 2002;40:4304-4307.
62. Rosato EA, Craig A, William, Archer LG. Quantitation of mecA transcription in oxacillin-resistant Staphylococcus aureus clinical isolates. J Bacteriol. 2003;185:3446–3452.
63. Volkmann H, Schwartz T, Bischoff P, Kirchen S, Obst U. Detection of clinically relevant antibiotic resistance genes in municipal wastewater using real-time PCR (TaqMan). J Microbiol Methods. 2004;56:277 286.
64. Theis T, Skurray AR, Brown HM. Identification of suitable internal controls to study expression of a Staphylococcus aureus multidrug resistance system by quantitative real-time PCR. J Microbiol Methods. 2007;70:355–362.
65. Diederen B, Kluytmans J, Vandenbroucke CM, Peeters MF. Utility of Real-Time PCR for Diagnosis of Legionnaires’ Disease in Routine Clinical Practice. J Clin Microbiol. 2008;46:671–677.
66. Stolhaug A, Bergh K. Identification and differentiation of Legionella pneumophila and Legionella spp. with real-time PCR targeting the 16S rRNA gene and species identification by mip sequencing. Appl. Environ. Microbiol. 2006;72:6394–6398.
67. Templeton KE, Scheltinga SA, Graffelman AW, Van Schie JM, Crielaard JW, Sillekens P, et al. Comparison and evaluation of real-time PCR, real time nucleic acid sequence-based amplification, conventional PCR, and serology for diagnosis of Mycoplasma pneumoniae. J. Clin. Microbiol. 2003;41:4366–4371.
68. Michelow IC, Olsen K, Lozano J, Rollins NK, Duffy LB, Ziegler T, et al. Epidemiology and clinical characteristics of communityacquired pneumonia in hospitalized children. Pediatrics. 2004;113:701–707.
69. Gullsby K, Storm M, Bondeson K. Simultaneous Detection of Chlamydophila pneumoniae and Mycoplasma pneumoniae by Use of Molecular Beacons in a Duplex Real-Time PCR. J Clin Microbiol. 2008;46:727–731.
70. Welti, Jaton MK, Altwegg M, Sahli R, Wenger A, Bille J. Development of a multiplex real-time quantitative PCR assay to detect Chlamydia pneumoniae, Legionella pneumophila and Mycoplasma neumoniae in respiratory tract secretions. Diagn Microbiol Infect. Dis. 2003;45:85–95.